PreSENTA, Evartia, Rodinia Oreana
【乳がんと卵巣がんのテスト】PreSENTIA BRCA1 / BRCA2 PANEL
はじめに
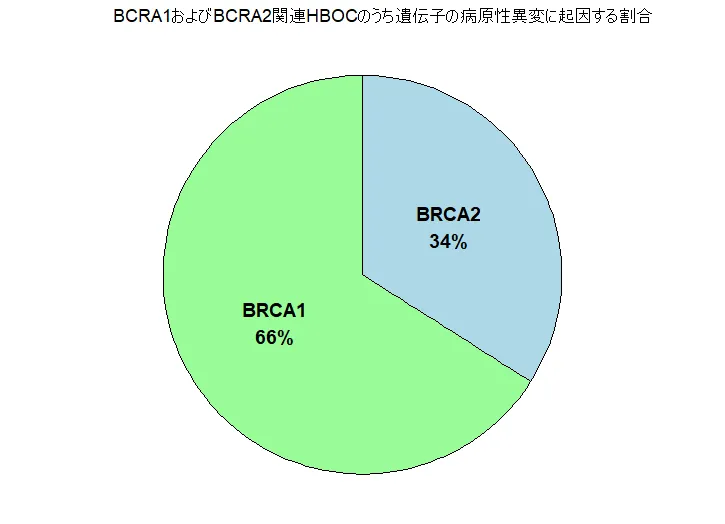
BRCA1およびBRCA2遺伝子の変異に関連する遺伝性乳がん・卵巣がん(Hereditary Breast and Ovarian Cancer:HBOC)は、乳がんや卵巣がんの発症リスクを大幅に高める遺伝性の疾患です。男女ともに影響を受ける可能性があり、これらの遺伝子に変異があると、乳がんや卵巣がんだけでなく、前立腺がん、膵臓がん、さらには皮膚がんの一種であるメラノーマなど、他のがんのリスクも上昇します。どのがんに対するリスクが特に高くなるかは、関与する遺伝子によって異なります。たとえば、BRCA1の変異は卵巣がんや若年性乳がんと強く関連しており、BRCA2の変異は男性乳がん、前立腺がん、膵臓がん、メラノーマとの関連がより高いことが分かっています。
HBOCの診断には、BRCA1またはBRCA2遺伝子に病的変異(Pathogenic Variant)があるかどうかを調べる遺伝子検査が必要です。この検査は、50歳未満で乳がんを発症した人や、男性乳がん、卵巣がんを発症した人、あるいは家族に同じようながんを発症した人が複数いる場合に推奨されます。一般的には、まず家族の中でがんを発症した人を検査し、その家系に特有の遺伝子変異を特定することから始めます。特に、アシュケナージ系ユダヤ人の家系では特定のBRCA変異が高頻度で見られるため、こうした人々には創始者変異(Founder Mutation)に焦点を当てた検査が推奨されることがあります。
HBOCの管理では、すでに発症したがんの治療に加えて、将来的ながんリスクを低減するための対策も重要です。乳がんに対しては、両側乳房切除術(Bilateral Mastectomy)と呼ばれる手術が、再発リスクを低減する選択肢の一つとして検討されます。また、BRCA変異を持つがん細胞に特有の性質を利用した治療として、PARP阻害剤(PARP Inhibitors)が乳がん、卵巣がん、前立腺がん、膵臓がんの治療に有効である可能性が示されています。さらに、乳房や卵巣、卵管を切除する手術ががんの予防策として有効であることも確認されています。たとえば、予防的両側乳房切除術や卵巣卵管摘出術(Salpingo-Oophorectomy)により、乳がんや卵巣がんのリスクを大幅に低下させることができます。また、卵管のみを先に切除し、閉経後に卵巣を摘出する方法も、早期閉経の影響を軽減しながらがん予防を目指す戦略として研究が進められています。
薬を用いた予防(化学予防:Chemoprevention)としては、タモキシフェンなどの薬剤が乳がんのリスクを低減する可能性があるとされています。ただし、これらの薬剤には血栓症や子宮内膜がんのリスクを増加させる可能性があるため、慎重な判断が求められます。また、経口避妊薬(Oral Contraceptives)は、BRCA変異を持つ女性において卵巣がんのリスクを下げる一方で、乳がんのリスクをわずかに高める可能性があると報告されています。さらに、HBOCに関連するがんを早期に発見するためには、定期的なスクリーニング(検査)が欠かせません。BRCA変異を持つ女性は、一般の人よりも若いうちから、マンモグラフィや乳房MRIによる乳がん検診を受けることが推奨されます。卵巣がんのスクリーニングには、経膣超音波検査(Transvaginal Ultrasound)や血液検査でCA-125という腫瘍マーカーを測定する方法がありますが、これらの検査だけでは卵巣がんを早期に確実に発見できるとは限らないため、慎重に判断する必要があります。男性の場合は、乳がんや前立腺がんのスクリーニングを行うことが推奨され、その他のがん(メラノーマや膵臓がんなど)については、個人や家族の病歴に基づいて検査方針が決められます。
HBOCと向き合ううえで、遺伝カウンセリング(Genetic Counseling)は非常に重要です。このカウンセリングでは、遺伝子検査の選択肢や結果の意味、がん予防や治療の選択肢について詳しく説明を受けることができます。HBOCは常染色体顕性遺伝(Autosomal Dominant Inheritance)で受け継がれるため、BRCA変異を持つ親の子どもは50%の確率で同じ変異を受け継ぐ可能性があります。ただし、BRCA変異を持つすべての人が必ずしもがんを発症するわけではなく、発症リスクは年齢、家族歴、生活習慣などさまざまな要因によって左右されます。現在も研究が進められており、BRCA1やBRCA2のどの変異が乳がんや卵巣がんのリスクをより高めるのか、また、リスクの高い人に対して最も効果的な管理方法は何かといった点が明らかになりつつあります。特に、PARP阻害剤などの新しい治療法は、BRCA変異を持つがん細胞の特性を活かした個別化医療(Personalized Medicine)の発展を象徴するものとなっています。
HBOCは、遺伝的要因と環境要因が複雑に絡み合って発症する疾患であり、その診断や管理には、個々の状況に応じたきめ細やかなアプローチが求められます。遺伝子検査の活用や新しい治療法の開発が進むことで、BRCA変異を持つ人々やその家族にとって、より早期の対策と適切な意思決定が可能になりつつあります。
BRCA1

乳がんは世界中で最も一般的ながんの一つで、新たに診断される全がんの約11.7%を占めています。その発症にはさまざまな要因が関与しますが、遺伝子の変異が大きく影響しています。特に「BRCA1(ビーアールシーエーワン)」という遺伝子に変異があると、乳がんや卵巣がんの発症リスクが大幅に高まることが知られています。
BRCA1遺伝子は、第17染色体上に位置しており、「乳がん感受性タンパク質1(breast cancer type 1 susceptibility protein)」という重要なタンパク質を作り出します。このタンパク質は、DNAの損傷を修復したり、細胞の増殖を適切に調節したりする働きを持ち、細胞が正常に機能するために不可欠です。もしBRCA1遺伝子に変異があると、この修復機能が十分に働かず、遺伝子の異常が蓄積してがんが発生しやすくなります。
BRCA1タンパク質は「腫瘍抑制因子」として機能し、がんの発生を防ぐ役割を果たします。このタンパク質は、他のタンパク質とともに「BRCA1関連ゲノム監視複合体(BASC)」と呼ばれるシステムを形成し、特にDNAの二本鎖切断を修復する重要な役割を担います。DNAの損傷が適切に修復されないと、細胞の異常な増殖につながり、がんが発生しやすくなります。
BRCA1遺伝子の変異は、特に「遺伝性乳がん」と深く関係しています。遺伝性乳がんとは、家族内でがんが発症しやすいケースを指し、BRCA1遺伝子に変異があると、乳がんの発症リスクは生涯で約80%、卵巣がんの発症リスクは40~65%に達するとされています。特に、BRCA1の変異は「トリプルネガティブ乳がん(TNBC)」というタイプの乳がんと関連が深いことが分かっています。TNBCは、ホルモン療法の対象となるエストロゲン受容体やプロゲステロン受容体を持たず、HER2タンパク質の異常増加も見られないため、治療が難しいタイプの乳がんです。
また、BRCA1遺伝子の変異は「家族性乳がん・卵巣がん症候群(BROVCA1)」とも関連しており、この症候群を持つ家系では、若い年齢で乳がんを発症しやすく、両側の乳房にがんが発生するケースや男性乳がんのリスクも高まることが知られています。さらに、膵臓がんや大腸がんなど、他のがんのリスクも増加することが報告されています。
BRCA1の機能低下は、遺伝子の変異だけでなく、エピジェネティックな変化(遺伝子の発現を制御する仕組みの変化)によっても引き起こされることがあります。例えば、BRCA1のプロモーター領域(遺伝子のスイッチのような部分)がメチル化されると、BRCA1の発現が抑えられ、散発的に乳がんが発生することがあります。また、一部のBRCA1が欠損したがん細胞は、DNAの再編成を通じてBRCA1の発現を回復し、PARP阻害剤やプラチナ系抗がん剤に対する耐性を獲得することが分かっています。このようながん細胞の適応力の高さは、治療の難しさにつながるため、新しい治療戦略の開発が求められています。
BRCA1の発見は、がんの遺伝子検査や個別化医療(患者ごとに最適な治療を選択する医療)の発展に大きく貢献しました。現在、BRCA1遺伝子の変異を検出する検査が広く実施され、高リスクの人に対して、定期的な検診や予防手術、薬によるがん予防の選択肢が提供されています。また、BRCA1変異がある場合、特定の治療薬が効果的であることが分かっており、例えば、プラチナ系抗がん剤やPARP阻害剤は、DNA修復の仕組みが弱まったがん細胞に対して特に有効とされています。
最近の研究では、BRCA1変異を持つ人の乳腺組織で、がんが発生する前にどのような変化が起こるのかが詳しく調べられています。研究結果によると、BRCA1やp53(がん抑制に関与する遺伝子)の変化が、乳腺細胞の異常な分化を引き起こし、免疫環境にも影響を与えることで、がんが発生しやすい状態になることが示唆されています。このような初期段階の変化を理解することは、新しい診断法や予防法の開発につながる可能性があり、乳がんや卵巣がんの発症リスクを下げるための手がかりとなると期待されています。
まとめると、BRCA1は乳がんや卵巣がんの発症に深く関わる重要な遺伝子であり、その機能はDNA修復やがんの抑制、細胞の正常な維持に欠かせません。遺伝子検査の普及や標的治療の発展により、BRCA1変異を持つ人のがん予防や治療の選択肢は大きく広がりました。しかし、がんの治療抵抗性(薬が効きにくくなる現象)や新たな適応メカニズムといった課題も残されており、今後もより効果的な治療法の開発が求められています。
BRCA2

BRCA2遺伝子の変異によって引き起こされる遺伝性乳がん卵巣がん症候群(Hereditary Breast and Ovarian Cancer: HBOC)は、乳がんや卵巣がんの発症リスクを高める遺伝的要因の一つとして重要視されています。BRCA2遺伝子は13番染色体の長腕(13q13.1)に位置し、「乳がん2型感受性タンパク質(Breast Cancer Type 2 Susceptibility Protein)」を作る働きを持っています。このタンパク質は、DNAの損傷を修復し、遺伝情報の安定性を保つ役割を担っています。特に、「相同組換え」と呼ばれるDNAの正確な修復メカニズムに関与し、細胞が正常に機能するために不可欠なものです。
BRCA2タンパク質は、DNA修復において中心的な役割を果たすRAD51という分子と連携し、DNAの損傷部分に適切に結合させることで修復を助けます。この過程では、DNAに結合しているレプリケーションタンパク質A(Replication Protein A: RPA)を置き換え、修復を安定化させます。また、PALB2やその他の相同組換えに関わるタンパク質とも協力しながら、DNAの複製が正確に行われるよう働きます。さらに、BRCA2は、遺伝情報が正しく転写される過程で生じる「Rループ」と呼ばれる不安定な構造を制御し、不要なDNA損傷を防ぐ役割も持っています。しかし、BRCA2遺伝子に変異が生じると、この修復機能が低下し、DNA損傷が蓄積することで遺伝情報が不安定になります。その結果、細胞のがん化が進み、がんの発症リスクが高まります。
BRCA2遺伝子の変異によって発症するHBOCは、正式には「家族性乳がん卵巣がん2型(Familial Breast-Ovarian Cancer Type 2: BROVCA2)」と呼ばれます。この疾患の特徴として、50歳以前の比較的若い年齢で乳がんや卵巣がんを発症しやすいことが挙げられます。具体的には、BRCA2変異を持つ女性の乳がん発症リスクは80歳までに約69%、卵巣がんの発症リスクは約17%と推定されています。また、BRCA2変異は乳がんや卵巣がんだけでなく、前立腺がんや膵臓がんのリスクを高めることも知られています。
BRCA2に関連するがんは、他の遺伝的要因によるがんと比べて特有の特徴を示すことが多いです。例えば、BRCA2変異を持つ家系では、男性の乳がんや両側乳がん(左右の乳房にそれぞれ独立して発生するがん)が比較的多く見られます。また、卵巣がんや前立腺がん、膵臓がんの発症率が高いことも、この遺伝子変異が多様な臓器に影響を及ぼすことを示しています。
BRCA2変異は非常に多様であり、遺伝子の一部が欠失したり、余分なDNA配列が挿入されたりすることで、正常なタンパク質が作られなくなるケースが多く見られます。こうした変異の影響を理解するために、さまざまな集団研究が行われており、BRCA2変異の遺伝的特性やその影響についての知見が蓄積されています。BRCA2変異は「常染色体優性遺伝」として受け継がれ、片方の親から変異した遺伝子を受け継ぐだけで、がんのリスクが上昇します。BRCA1とBRCA2の両方がHBOCの原因となりますが、それぞれのがんリスクには違いがあります。例えば、卵巣がんの発症リスクはBRCA1変異を持つ人の方が高い傾向がありますが、男性乳がんや膵臓がんのリスクはBRCA2変異を持つ人の方が高いとされています。
さらに、遺伝的要因だけでなく、環境要因やライフスタイルもBRCA2変異を持つ人のがんリスクに影響を与えます。一般的に、妊娠回数が多いことは乳がんリスクを低下させる要因の一つとされていますが、BRCA2変異を持つ人にとっては、必ずしも同じような保護効果があるとは限りません。一部の研究では、特定の条件下では妊娠が乳がんリスクをわずかに増加させる可能性が示唆されています。
近年の遺伝子検査技術の進歩により、BRCA2変異の有無を特定し、一人ひとりのがんリスクをより正確に評価することが可能になっています。特に、アシュケナージ系ユダヤ人、アイスランド人、フランス系カナダ人など、特定の集団ではBRCA2変異が比較的高い頻度で見られるため、これらのグループに属する人や、家族内に乳がんや卵巣がんの発症歴がある人には遺伝子検査が強く推奨されます。BRCA2変異が確認された場合、がんの早期発見のためのスクリーニングを強化したり、予防的な手術を検討したりすることが可能です。また、最近では、BRCA2欠損がんの特徴を利用した治療法の研究が進んでおり、例えば、PARP阻害薬という分子標的治療が有効であることが明らかになっています。
BRCA2変異に関する研究は現在も進行中であり、がんリスクに影響を与える他の遺伝的要因についても新たな知見が得られつつあります。例えば、FGFR2、MAP3K1、TOX3といった遺伝子の特定の変異が、BRCA2変異を持つ人の乳がんリスクに関与していることが報告されています。こうした研究は、遺伝的要因の複雑な相互作用を明らかにするだけでなく、個別化されたリスク評価や治療の可能性を広げることにつながります。
BRCA2はDNA修復の要となる遺伝子であり、その変異は遺伝性乳がんや卵巣がんの大きな要因となります。分子メカニズム、集団遺伝学、臨床的な特徴についての理解が深まることで、BRCA2変異を持つ人々の予防や治療の選択肢が拡充されています。特に、がんの早期発見と適切な対策が重要であり、遺伝子検査を活用することで、発症前に適切な対応を取ることが期待されます。
【代謝病のテスト】EVARTIA Comprehensive Metabolic Genetic Testing
はじめに
Evartiaの代謝遺伝子検査は、遺伝性代謝疾患に関連する遺伝子変異を特定するために設計された、包括的かつ最先端の検査ツールです。この検査では、代謝経路に関与する223種類の遺伝子を対象とした詳細なパネルを解析し、複雑な疾患の検出を簡略化します。また、当社の専門家によって開発された高度な「ターゲットキャプチャーエンリッチメント技術(Target Capture Enrichment Technology)」を基盤としています。この技術により、効率的かつ正確な解析を可能にしています。
遺伝性代謝疾患とは
遺伝性代謝疾患は、代謝に関与する遺伝子の異常(欠損や変異)が原因で発生します。これらの異常は代謝経路の働きを妨げ、有害物質の蓄積や重要な酵素・タンパク質の不足を引き起こします。その結果、症状は新生児期に急性かつ生命を脅かす形で現れる場合もあれば、成人期にゆっくり進行しながら顕在化する場合もあります。さらに、食事、病気、脱水症状といった要因によって症状が誘発されることもあり、迅速かつ正確な診断が求められます。
Evartiaの特徴
Evartiaは、特に成人の診断において課題となる代謝疾患の発見を支援します。代謝疾患は、神経学的、精神的、心血管系の疾患と症状が重複することが多く、診断が困難です。しかし、Evartiaは精密な遺伝子検査を通じて、病気の根本原因を迅速に特定し、患者ごとに最適化された治療法の選択を可能にします。これにより、症状の改善や疾患の進行抑制、さらには生活の質(QOL)の向上に貢献します。
この検査が特に役立つケース
- 原因不明の持続的な症状を持つ患者や、代謝疾患の家族歴がある方
- 標準治療では効果が見られない神経学的症状を持つ方
- 幼少期から成人期まで、発症年齢や症状が多様なケース
次世代シーケンシング技術(NGS:Next-Generation Sequencing)
Evartiaは、次世代シーケンシング技術を活用することで、単一塩基多型(SNV:Single Nucleotide Variants)、小規模な挿入・欠失(Indel:Insertions and Deletions)、およびコピー数変異(CNA:Copy Number Alterations)を検出します。このように幅広い解析を行うことで、包括的な結果を提供します。さらに、検査結果は米国医科遺伝学会(ACMG:American College of Medical Genetics and Genomics)の基準に基づいて分類され、臨床管理に活用できる信頼性の高い情報を提供します。
Evartiaの意義
Evartiaは、侵襲的な検査への依存を軽減し、患者やその家族に早期診断の機会を提供します。この検査を通じて、患者と医療提供者は、的確でタイムリーな意思決定を行うことができ、人生を大きく変える治療の道を切り開くことができます。
遺伝性代謝疾患の概要
3-メチルグルタコン酸尿症(3-Methylglutaconic aciduria)
| REPRESENTATIVEDISEASES (JA) | バース症候群; 3-メチルグルタコン酸尿症 3 型、3-メチルグルタコン酸尿症 5 型 |
|---|---|
| REPRESENTATIVEDISEASES (EN) | Barth Syndrome; 3-Methylglutaconic aciduria Type 3; 3-Methylglutaconic aciduria Type 5 |
| IMPLICATEDGENES | AUH, CLPB, DNAJC19, OPA3, SERAC1, TAFAZZN, TIMM50, TMEM70 |
バース症候群(Barth Syndrome)
バース症候群(Barth Syndrome, BTHS)は、X連鎖性遺伝子疾患の一つで、主に男性に発症し、身体の複数のシステムに深刻な影響を及ぼします。この疾患は1983年に初めて記載され、臨床的には心筋症(cardiomyopathy, CMP)、骨格筋の弱さ、成長の遅れ、好中球減少症(neutropenia、白血球の一種である好中球の数が減少する状態)、および尿中3-メチルグルタコン酸(3-methylglutaconic acid, 3-MGCA)の増加といった特徴が組み合わさることで知られています。これまでに世界で報告された症例は200例未満と非常に稀な疾患ですが、その広範かつ多様な症状により診断が見逃されている可能性があると考えられています。
バース症候群の特徴的な症状は心筋症で、これが幼少期に発症することが多いです。患者の多くは5歳までに症状を呈し、その大半は乳児期に発症します。中でも拡張型心筋症(dilated cardiomyopathy, DCM)が最も一般的ですが、肥大型心筋症(hypertrophic cardiomyopathy, HCM)や左室非コンパクション症(left ventricular non-compaction, LVNC)も頻繁に見られます。さらに、心内膜線維弾性症(endocardial fibroelastosis, EFE)や心室性不整脈が心臓の問題を複雑化させます。多くの患者は標準的な心不全治療に良好に反応しますが、一部の患者は症状が進行し、約14%が心臓移植を必要とします。QT延長症候群(prolonged QT interval)や突然死も重大なリスクであり、早期診断と定期的な心臓のモニタリングの重要性を強調しています。
心臓の問題に加えて、バース症候群は骨格筋の弱さや運動発達の遅れを伴い、これが疲労感や体力の低下に繋がります。また、成長の遅れや思春期の遅延が一般的であり、多くの乳児は授乳困難、発育不良、反復性の下痢を経験します。好中球減少症は断続的かつ軽度な場合もあれば、持続的で重篤な場合もあり、細菌感染や敗血症への罹患リスクを高めます。興味深いことに、バース症候群の患者の中には尿中3-MGCAの増加や好中球減少症を示さない例もあり、この疾患の多様性と診断の難しさが浮き彫りになります。
バース症候群の根本的な原因は、X染色体のXq28に位置するTAZ遺伝子の変異や欠失です。この遺伝子はタファジン(tafazzin)というタンパク質をコードしており、ミトコンドリアの内膜に存在する主要なリン脂質であるカルジオリピン(cardiolipin)の再構築に重要な役割を果たします。カルジオリピン再構築の障害は細胞内のエネルギー産生に影響を与え、この疾患が多系統にわたる症状を呈する原因となります。診断法の進歩により、カルジオリピン分析や遺伝子検査を用いた迅速で正確なバース症候群の診断が可能となりました。これらの検査は、血液や保存された組織、さらには新生児の血液スポットなど、さまざまな生体試料で実施でき、患者の診断だけでなく、女性の保因者検出や出生前スクリーニングにも役立っています。
歴史的に、バース症候群は主に心疾患として認識されていましたが、現在では多系統性疾患として理解されており、産科医、小児科医、心臓専門医、神経内科医など、多くの専門医が最初に診る可能性があります。この疾患の症状の幅広さにより、好中球減少症や心筋症といった典型的な特徴が見られない場合には診断が難しくなることがあります。また、バース症候群は胎児死亡、男性の反復性流産、死産の原因である可能性も指摘されており、医療従事者の認識を高めることが重要です。
バース症候群の管理は複雑であり、多職種のチームアプローチが必要です。治療は患者ごとの症状に焦点を当てます。心臓の問題には標準的な心不全治療が一般的に使用され、一部の患者は機械的補助装置や移植などの高度な治療から恩恵を受けます。好中球減少症には予防的抗生物質や、重症例では顆粒球コロニー刺激因子(granulocyte colony-stimulating factor, G-CSF)療法を用いて白血球の生成を促進します。専門的なクリニックでの定期的なモニタリングと包括的なケアにより、多くの患者が成人期に達し、より長く健康的な生活を送れるようになっています。
バース症候群の早期発見は極めて重要であり、適時の診断により適切な管理が可能となるだけでなく、家族のスクリーニングや遺伝カウンセリングも実現します。原因不明の拡張型心筋症や左室非コンパクション症、説明のつかない心室性不整脈、突然死、または持続的な好中球減少症を持つ男児には、バース症候群の検査を検討すべきです。さらに、原因不明の成長遅延、授乳困難、反復性細菌感染、またはミトコンドリア疾患が疑われる場合、特にX連鎖性遺伝の家族歴がある場合には、この疾患を考慮する必要があります。
かつては極めて稀な疾患とされていたバース症候群ですが、認知の向上と診断技術の進歩により、その広範な臨床スペクトラムが明らかになりつつあります。医療従事者、研究者、患者支援団体が連携して研究を続けることで、診断率の向上、治療法の拡充、そしてこの困難な疾患を抱える人々の生活の質の向上が期待されています。
3-メチルグルタコン酸尿症 3 型(3-Methylglutaconic aciduria Type 3)
3-メチルグルタコン酸尿症タイプIII、別名コステフ視神経萎縮症候群または視神経萎縮タイプ3(Optic Atrophy Type 3, OPA3)は、非常に稀な遺伝性疾患であり、19番染色体上のOPA3遺伝子の変異によって引き起こされます。この疾患は常染色体劣性遺伝疾患の一種であり、ミトコンドリア機能に影響を及ぼすことが特徴です。この結果、主に視神経と運動機能に影響を与える神経眼科的な症状が現れます。
この病気の主な症状は、幼少期に発症する両側性の視神経萎縮、つまり視神経の損傷による視力低下です。これが最初に認められる症状であり、進行性の視力障害を引き起こします。その後、筋肉の硬直を伴う痙性や、不随意運動といった錐体外路症状が現れることが多く、場合によっては認知機能が低下するケースもあります。ただし、こうした神経症状が進行しても、多くの場合、患者の寿命には大きな影響を与えないとされています。
この疾患の生化学的特徴として、尿中に特定の有機酸が増加して排泄される点が挙げられます。特に、3-メチルグルタコン酸と3-メチルグルタル酸という化合物が顕著に増加します。これらはロイシンというアミノ酸の代謝や、メバロン酸シャント経路と呼ばれる体内の特定の代謝経路に関与する中間生成物です。尿中のこれらの有機酸濃度の上昇は、3-メチルグルタコン酸尿症タイプIIIを特定するための重要な診断指標となります。特に幼少期に視神経萎縮が認められた場合、この指標を用いることで早期診断が可能です。
この疾患は、もともとイラク系ユダヤ人の集団で発見されました。この集団では、OPA3遺伝子に特定の変異(IVS1-1G>C)が存在しており、これが疾患の原因であることが分かりました。この変異は、細胞内でmRNAの発現を妨げ、OPA3タンパク質の欠乏を引き起こします。OPA3遺伝子は、179アミノ酸から構成されるタンパク質をコードしており、このタンパク質は主にミトコンドリアに局在します。ミトコンドリアへの局在は、タンパク質内のリーダー配列や輸送シグナルによって誘導されます。OPA3タンパク質のミトコンドリア内での役割は完全には解明されていませんが、疾患に関連する代謝異常がミトコンドリアの機能不全に起因していることを裏付ける証拠となっています。
この病気は特定の民族集団に多いとされていましたが、他の集団でも同様の症例が報告されています。例えば、クルド系トルコ人の患者でOPA3遺伝子変異が確認された例もあり、これにより3-メチルグルタコン酸尿症タイプIIIが特定の民族に限らない疾患であることが明らかになりました。
3-メチルグルタコン酸尿症は、尿中に3-メチルグルタコン酸や3-メチルグルタル酸が排泄される代謝異常症の一つであり、大きく4つのタイプに分類されます。タイプIは、ロイシン分解に関与する酵素である3-メチルグルタコニル-CoAヒドラターゼの欠乏によるもので、軽度の神経症状を引き起こします。タイプIIはバース症候群と呼ばれるX連鎖性疾患で、tafazzin遺伝子の変異によって心筋症や好中球減少症などの症状が現れます。タイプIVは、神経や心臓、肝臓、腎臓に多様な症状が現れる異質な疾患群です。その中で、タイプIIIはOPA3遺伝子変異により引き起こされ、ミトコンドリア機能不全が明確に関連する唯一のタイプであるとされています。
このように、3-メチルグルタコン酸尿症タイプIIIは視力低下から始まり、運動機能や代謝異常が続いて現れる特徴的な疾患です。特に幼少期の視力障害が早期診断の重要な手がかりとなるため、初期症状が現れた場合は適切な検査を受けることが推奨されます。
3-メチルグルタコン酸尿症 5 型(3-Methylglutaconic aciduria Type 5)
3-メチルグルタコン酸尿症5型(MGCA5)は、「拡張型心筋症と運動失調を伴う症候群(DCMA症候群)」としても知られる、非常に稀な常染色体劣性遺伝性疾患です。この病気は、3q26染色体上に位置するDNAJC19遺伝子の変異によって引き起こされます。この遺伝子は、ミトコンドリア内で特定のタンパク質の正常な機能を維持するための「シャペロンタンパク質」をコードしています。MGCA5はカナダのダリウスルート・ハッタライト集団(宗教的少数派グループ)で初めて確認され、この集団内では特定のDNAJC19遺伝子変異が報告されています。
MGCA5の主な特徴は、乳児期や幼児期に発症する拡張型心筋症(DCM)です。拡張型心筋症は、心臓が弱く拡大し、血液を効率的に送り出せなくなる状態です。さらに、一部の患者では、左室非コンパクションという心筋の異常が見られます。これは、胎児期の心筋発達が不完全なために、心筋がスポンジ状に見える構造的異常です。また、患者の多くは長QT症候群(心電図上でQT間隔が延長する状態)を発症し、不整脈、失神、重篤な場合には心停止や突然死を引き起こすリスクがあります。特に心不全は、幼少期における死亡の主要な原因となっていますが、心臓に関連する症状を示さない患者も一部存在することから、この病気の症状には個人差があることが分かります。
心臓以外の症状としては、成長障害が挙げられます。この成長障害は出生前後の段階から認められ、最終的に低身長につながることが多いです。運動機能の問題も一般的で、特に運動失調(体のバランスや協調性の障害)は2歳頃までに現れることが多いです。これにより歩行などの発達が遅れる場合もありますが、多くの子どもは成長とともに自立歩行が可能になります。知的障害は軽度の場合が多いものの、個人差があります。
男性患者には、停留精巣(精巣が陰嚢内に下降しない状態)や尿道下裂(尿道の開口部が正常位置にない状態)などの生殖器異常が見られることが一般的です。また、小球性貧血(通常より小さい赤血球を特徴とする貧血)もよく認められ、これにより皮膚が青白く見えたり、疲労感が生じたりします。さらに、視神経萎縮(視覚信号を脳に伝達する視神経の変性)による視力低下や、肝臓に脂肪が蓄積する脂肪肝(肝脂肪変性)も報告されています。
MGCA5を診断するための重要な指標として、尿中の3-メチルグルタコン酸と3-メチルグルタル酸の濃度が挙げられます。これらの物質はミトコンドリアの機能障害により蓄積します。尿検査によるこの特徴的な代謝異常の確認は、遺伝子検査を行う前にMGCA5を他の代謝性疾患と区別する上で重要です。ただし、これらの酸の異常値が病気の重症度や進行に直接影響を及ぼすわけではありません。
MGCA5は、拡張型心筋症や3-メチルグルタコン酸尿症を特徴とする別のミトコンドリア病「バース症候群」と一部の症状が似ています。しかし、バース症候群はX連鎖性(X染色体に関連する遺伝形式)であり、MGCA5とは異なる筋肉の弱さや好中球減少症(白血球の一種の減少)などの特徴があります。
MGCA5患者30人以上を対象とした研究データによると、症状の発症年齢の中央値は生後12か月であり、生後1か月で発症した例から3歳で発症した例まで幅広く存在します。この研究では、全患者が成長障害を示し、67%が貧血、61%が拡張型心筋症、55%が運動失調または知的障害、22%が視神経萎縮、33%が長QT症候群を伴っていました。また、男性患者の多くに停留精巣や尿道下裂などの生殖器異常が見られました。悲しいことに、患者の約60%が心不全や不整脈といった合併症で亡くなっており、平均死亡年齢は22か月とされています。
このように診断が難しいケースも多いものの、MGCA5の遺伝的および生化学的特徴の理解が進むことで、早期発見と治療の可能性が高まっています。心筋症や尿中代謝異常、全身性の症状を組み合わせて検討することで、他の小児心筋症との鑑別が可能となります。しかし、症状の重症度や範囲には個人差が大きく、それぞれの患者に応じた個別ケアが必要であることが示唆されています。
脳クレアチン欠乏症(Cerebral creatine deficiency)
| REPRESENTATIVEDISEASES (JA) | 脳クレアチン欠乏症候群 1; 脳クレアチン欠乏症候群 2 |
|---|---|
| REPRESENTATIVEDISEASES (EN) | Cerebral creatine deficiency syndrome 1; Cerebral creatine deficiency syndrome 2 |
| IMPLICATEDGENES | GAMT, GATM, SLC6A8 |
脳クレアチン欠乏症候群 1(Cerebral creatine deficiency syndrome 1)
脳クレアチン欠乏症1型(Cerebral Creatine Deficiency Syndrome 1, CCDS1)は、主に脳の機能に影響を及ぼすまれな遺伝性疾患です。この病気はX染色体上の遺伝子異常によって引き起こされるため、「X連鎖性疾患」と呼ばれます。男性はX染色体を1本しか持たないため、遺伝子異常の影響を直接受けやすく、症状が重くなる傾向があります。CCD1の有病率は男性で0.3%〜3.5%と推定されています。一方、女性はX染色体を2本持っているため、片方の遺伝子に異常があっても通常は比較的軽い症状しか現れず、「保因者」として次世代に遺伝する可能性があります。しかし、一部の女性保因者にも軽度の神経学的な問題がみられることがあります。
この疾患は、SLC6A8遺伝子の変異によって引き起こされます。この遺伝子はX染色体のXq28領域に位置し、クレアチンの輸送を担うタンパク質を作る役割を持っています。クレアチンは、脳や筋肉の細胞がエネルギーを蓄え、効率的に利用するために不可欠な物質です。しかし、SLC6A8遺伝子に変異があると、クレアチンを細胞に取り込む働きが正常に機能しなくなり、結果として脳の発達や機能にさまざまな問題が生じます。
CCDS1の症状は乳児期から幼児期にかけて現れることが多く、最も顕著なのは知的発達の遅れです。その程度は軽度から重度までさまざまで、特に言語の発達が大幅に遅れるのが特徴です。中には言葉を使ったコミュニケーションがほとんどできない場合もあります。また、多くの子どもに注意欠陥・多動性障害(ADHD)や自閉スペクトラム症(ASD)の特徴がみられ、社会的なやりとりや感情のコントロールが難しいことがあります。
さらに、CCDS1の患者にはてんかん発作がよくみられますが、その頻度や重症度は個人によって異なります。また、運動発達の遅れも一般的で、通常よりも座る、這う、歩くといった基本的な動作を習得するのが遅れることが多いです。加えて、疲れやすく、持久力が低いという特徴もあります。
CCDS1の患者の中には、心臓のリズム異常(不整脈)を伴うケースもあり、まれに突然死のリスクが指摘されています。また、一部の患者には小頭症(年齢に比べて頭が小さいこと)がみられ、それに伴い発達の遅れが生じることもあります。さらに、顔の特徴が独特になる場合があり、たとえば額が広い、または顔の中央部分がへこんだように見える(中顔面低形成)といった特徴がみられることもあります。
CCDS1はX連鎖性劣性遺伝形式で遺伝するため、女性保因者は通常、重症化することはありません。しかし、一部の女性は軽度の学習障害や行動面での変化を経験することがあります。
CCDS1の診断は、遺伝子検査によって確定されます。この検査では、SLC6A8遺伝子の変異を特定することで診断が可能です。また、磁気共鳴スペクトロスコピー(MRS)という脳の画像診断技術を用いると、脳内のクレアチン濃度が低下していることが確認される場合があります。
現在のところ、CCDS1を根本的に治療する方法は確立されていません。そのため、治療の中心は症状の管理になります。例えば、言語療法(スピーチセラピー)や理学療法(フィジカルセラピー)、行動療法を組み合わせることで、コミュニケーション能力や運動機能、社会的スキルの向上を目指すことが重要です。また、てんかん発作がある場合には抗てんかん薬が使用されることがあります。さらに、学習障害のある子どもには、特別支援教育などのサポートが提供されることもあります。
一般的にクレアチン補充療法は、体内でクレアチンが十分に作られない他の種類のクレアチン欠乏症には有効ですが、CCDS1の場合、問題の本質は「クレアチンの輸送障害」にあるため、クレアチンサプリメントの効果はほとんど期待できません。
CCDS1の研究は現在も進められており、将来的にはより効果的な治療法が開発される可能性があります。早期診断と適切な支援が、患者の生活の質を向上させる鍵となるため、この疾患についての理解を深めることが重要です。
脳クレアチン欠乏症候群 2(Cerebral creatine deficiency syndrome 2)
脳クレアチン欠乏症候群2(CCDS2)、またはグアニジノ酢酸メチル基転移酵素(GAMT)欠損症は、主に脳と筋肉に影響を及ぼすまれな遺伝性疾患です。この疾患は常染色体劣性遺伝疾患に分類され、発症には両親からそれぞれ1つずつ、変異したGAMT遺伝子を受け継ぐ必要があります。
GAMT欠損症は、クレアチン代謝に関連する最初に発見された疾患として知られています。この病気の原因は、クレアチン合成に不可欠な酵素であるグアニジノ酢酸メチル基転移酵素(GAMT)が不足することです。クレアチンは、特に脳や筋肉においてエネルギーを生産する重要な役割を果たしており、不足すると神経機能や発達に深刻な影響を及ぼします。
GAMT欠損症の症状は乳児期から幼児期(通常3歳まで)にかけて現れることが多く、その重症度や具体的な症状は個人によって異なります。しかし、一般的に見られる症状として、知的障害、発達の遅れ、著しい言語障害が挙げられます。特に言語発達の遅れが顕著で、話せる単語がごくわずかに限られるケースが多く報告されています。
また、てんかん(繰り返し起こる発作)が頻繁に見られ、発作のコントロールが難しい場合もあります。加えて、震え(振戦)、顔の筋肉のピクピクした動き(チック)、その他の不随意運動(自分の意思とは関係なく起こる動き)などの運動異常がみられることもあります。こうした運動障害は、脳の錐体路(すいたいろ)および錐体外路(すいたいがいろ)と呼ばれる神経回路の機能異常によるものと考えられています。
行動面においても問題が見られることが多く、多動傾向や自閉症的特徴(社会的なやりとりやコミュニケーションの困難)が報告されています。また、一部の子どもでは、頭を壁に打ち付けるなどの自傷行動をとることがあります。
運動発達の遅れも顕著で、座る、歩くといった基本的な運動機能の獲得が遅れるケースが多く見られます。重症の場合、すでに獲得した運動スキル(例:頭を支える、座る)が失われることもあります。また、筋緊張異常(特に筋肉の低緊張=筋力の弱さ)がみられ、運動のしづらさの原因となることがあります。
GAMT欠損症の特徴的な生化学的異常として、グアニジノ酢酸(GAA)が脳内および体液に蓄積する一方で、脳内のクレアチンおよびホスホクレアチンが著しく減少することが知られています。この代謝異常が、神経系の障害を引き起こす主な原因となります。
診断は、臨床症状に基づいて行われ、さらに検査によって確定されます。特に、尿中のグアニジノ酢酸(GAA)の濃度が高いことが重要な診断指標の一つです。加えて、GAMT遺伝子(染色体19p13に存在)の変異を特定する遺伝子検査を行うことで確定診断が可能になります。また、脳の磁気共鳴スペクトロスコピー(MRS)を用いた画像検査では、脳内のクレアチンの欠乏が確認されることがあります。
GAMT欠損症には治療法が存在し、早期に治療を開始すれば非常に効果的であることが知られています。治療の基本は、食事療法とクレアチンサプリメントの補充です。クレアチンを補給することで、脳内のクレアチン不足を改善し、症状の進行を抑えることが可能になります。また、アルギニンなどのグアニジノ酢酸の前駆物質(体内でGAAに変わる成分)の摂取を制限することで、有害な代謝物の蓄積を抑えることが推奨されています。
しかし、治療が遅れると、すでに生じた脳の損傷を回復させることは困難であり、知的障害や運動機能の異常が長期的に残る可能性があります。そのため、早期発見と早期治療が極めて重要です。
先天性糖鎖障害(Congenital disorders of glycosylation)
| REPRESENTATIVEDISEASES (JA) | 先天性糖鎖障害 Ic 型; 先天性糖鎖障害 Ia 型 |
|---|---|
| REPRESENTATIVEDISEASES (EN) | Congenital disorder of glycosylation type Ic; Congenital disorder of glycosylation type Ia |
| IMPLICATEDGENES | ALG1, ALG2, ALG3, ALG6, ALG8, ALG9, ALG11, ALG12, ALG13, B4GALT1, CAD, CCDC115, COG1, COG2, COG4, COG5, COG6, COG7, COG8, DDOST, DHDDS, DOLK, DPAGT1, DPM1, DPM2, DPM3, FUT8, GMPPA, GNE, MAN1B1, MGAT2, MOGS, MPDU1, MPI, NGLY1, NUS1, PGM1, PMM2, RFT1, SLC35A1, SLC35A2, SLC35C1, SLC39A8, SRD5A3, SSR4, STT3A, STT3B, TMEM165, TMEM199, TUSC3 |
先天性糖鎖障害 Ic 型(Congenital disorder of glycosylation type Ic)
先天性糖鎖異常症タイプIc(CDG1C)は、体のさまざまな機能に影響を及ぼすまれな遺伝性疾患です。この病気は、ALG6遺伝子(染色体1p31に位置)の変異によって引き起こされます。ALG6遺伝子は糖タンパク質の合成において重要な役割を担っており、特にN-結合型オリゴ糖(タンパク質に付加される糖鎖)の組み立て時に、マンノースという糖を適切に追加する過程に関与しています。このプロセスが正常に機能しないと、糖タンパク質の形成が阻害され、細胞の成長や分化、維持に支障をきたします。そのため、この疾患は複数の臓器や機能に影響を及ぼし、多様な症状を引き起こします。
CDG1Cの症状は乳児期から現れることが多く、患者ごとに大きな個人差があります。最も早期に見られる兆候の一つは発育不良(成長障害)で、赤ちゃんが十分に体重を増やせず、期待される成長速度に達しないことがあります。また、筋緊張低下(筋肉の力が弱い状態)がよく見られ、これにより首のすわりの遅れ、歩行の困難さ、言葉の発達の遅れといった運動発達の遅れが生じます。さらに、多くの子どもが神経系の異常を抱え、運動失調(バランスや協調運動の障害)やけいれん発作(熱性けいれんからてんかん発作まで幅広い)を経験します。一部の患者では、脳卒中様発作が発生し、突然の強い倦怠感や一時的な麻痺を引き起こすこともあります。
また、視覚の問題もCDG1Cの特徴の一つです。患者の多くは斜視(両目の焦点が合わない状態)を伴い、一部の患者では網膜色素変性症(進行性の視力障害を引き起こす疾患)が見られることがあります。加えて、消化器系に影響が及ぶこともあり、タンパク喪失性腸症(腸を通じて重要なタンパク質が失われ、栄養不足を引き起こす状態)を発症するケースもあります。肝臓の機能異常が見られる場合もありますが、その程度は患者によって異なります。さらに、血液凝固異常が現れることがあり、これにより出血しやすくなったり、逆に血栓ができやすくなったりするといったリスクが生じることがあります。
女性においては、CDG1Cは高ゴナドトロピン性性腺機能低下症(思春期の正常な発達を妨げるホルモン異常)と関連しています。そのため、多くの患者は思春期を迎えず、二次性徴が現れないという特徴があります。
CDG1Cは、より広範な疾患群である先天性糖鎖異常症(CDG: Congenital Disorders of Glycosylation)に分類されます。この疾患群は、かつて糖タンパク質欠乏症候群(CDGS: Carbohydrate-Deficient Glycoprotein Syndromes)と呼ばれていました。CDGは、糖鎖の形成過程のどの段階に異常があるかによって大きく2つのタイプに分類されます。CDGタイプI(CDG-I)は、糖鎖がタンパク質に付加される前の形成や転送に異常があるタイプで、CDG1Cはこのグループに属します。一方、CDGタイプII(CDG-II)は、糖鎖がタンパク質に付加された後の修飾過程に異常が生じるタイプです。
CDG1Cは、CDGの中でも2番目に多いサブタイプとされており、まれな疾患ではありますが、比較的よく知られている種類の一つです。CDG1Cは全身に影響を及ぼすため、診断には遺伝子検査や血液検査を行い、糖タンパク質の異常を確認することが一般的です。
現在のところ、CDG1Cを根本的に治療する方法はなく、治療の中心は症状の管理と生活の質の向上となります。運動発達の遅れに対しては理学療法や作業療法が行われ、けいれん発作には抗てんかん薬が使用されることがあります。また、栄養面のサポートとして適切な食事管理や栄養補給が必要となり、血液凝固異常やホルモンバランスの異常についても定期的にモニタリングすることが重要です。
症状の現れ方や重症度には個人差がありますが、早期診断と適切な介入によって、患者の生活の質を大きく向上させることが可能です。
先天性糖鎖障害 Ia 型(Congenital disorder of glycosylation type Ia)
先天性糖鎖異常症Ia型(CDG1A)は、PMM2遺伝子の変異によって引き起こされるまれな遺伝性疾患です。糖タンパク質の合成異常により、複数の臓器や機能に影響を及ぼします。この疾患は、PMM2(ホスホマンノムターゼ-2)酵素を作るための遺伝情報に異常があることが原因で発症します。PMM2酵素は、体内で重要な働きをするN-糖鎖付加(N-グリコシル化)というプロセスに関与し、糖鎖(グリカン)をタンパク質に結合させることで、その構造や機能を調整する役割を果たします。この糖鎖付加は、細胞間のシグナル伝達、代謝、免疫応答、組織の発達や維持などに不可欠な機能です。そのため、この過程に異常が生じると、さまざまな症状が現れます。
CDG1Aは、常染色体劣性遺伝の形式で遺伝します。つまり、両親からそれぞれ1つずつ変異したPMM2遺伝子を受け継いだ場合に発症します。同じ家族内でも症状の重さや現れ方には大きな違いがあり、患者ごとに異なる症状が見られます。
CDG1Aの症状は乳児期に現れることがほとんどです。代表的な初期症状には、筋緊張の低下(低緊張)、発達の遅れ、摂食障害による発育不良が挙げられます。また、多くの乳児には独特な顔貌が見られ、広い額、三角形の顔、目立つ耳の形、薄い上唇などの特徴を持つことがあります。さらに、乳首の陥没(反転乳頭)や異常な脂肪分布などもCDG1Aに特有の身体的特徴です。
CDG1Aにおける神経系の異常の特徴的な症状として、小脳低形成(小脳の発達不足)があります。小脳は運動の調整を担うため、この異常により運動機能の低下、協調運動障害(運動失調)、精神運動発達の遅れが生じます。また、多くの患者で眼球運動異常(斜視など)が見られ、進行性の視力障害(網膜色素変性症)を発症することもあります。その他、けいれん発作、知的障害、末梢神経障害(手足の感覚の低下や筋力低下)などがみられることもあります。
CDG1Aは神経系だけでなく、多臓器にも影響を及ぼします。一部の乳児では肝機能異常、心嚢水貯留(心臓周囲に液体がたまる状態)、血液凝固異常が認められます。これらの合併症のため、約20%の患者が1歳までに多臓器不全、重篤な感染症、または心筋症などで死亡すると報告されています。
最も重症なケースでは、胎児水腫(体内に過剰な水分が蓄積する状態)を引き起こすことがあり、この場合、ほとんどの赤ちゃんが生存できません。
乳児期を乗り越えた場合でも、多くの患者が中等度の知的障害を持ち、一部の患者は歩行が独立してできないことがあります。また、一過性の麻痺や極度の倦怠感を伴う脳卒中様エピソードを経験することがあり、通常は数週間から数ヶ月のうちに回復しますが、生活の質に大きな影響を及ぼします。
年齢を重ねると、新たな合併症が現れることがあります。進行性の末梢神経障害による手足の筋力低下や感覚異常がみられるほか、脊柱の異常(後弯・側弯)、関節拘縮(関節が硬くなり動きが制限される状態)が見られることもあります。
また、ホルモン異常もよくみられます。女性では高ゴナドトロピン性性腺機能低下症により思春期が正常に進行しないことが多いです。一方、男性は思春期を迎えるものの精巣が小さいという特徴があります。
CDG1Aは、先天性糖鎖異常症(CDG)の中で最も一般的なタイプであり、世界中で700〜800例以上の報告があります。しかし、診断されていない患者も多いと考えられ、実際の発症頻度はさらに高い可能性があります。
診断には遺伝子検査が行われ、PMM2遺伝子の変異を特定することで確定されます。また、血清トランスフェリン解析により糖鎖付加の異常を検出し、脳MRI検査では小脳低形成が確認されることが一般的です。
脂肪酸酸化障害(Fatty acid oxidation disorders)
| REPRESENTATIVEDISEASES (JA) | 中鎖アシルCoA脱水素酵素(MCAD)欠損症、短鎖アシルCoA脱水素酵素(SCHAD)欠損症、全身性一次性カルニチン欠損症、極長鎖アシルCoA脱水素酵素(VLCAD)欠損症 |
|---|---|
| REPRESENTATIVEDISEASES (EN) | Medium chain acyl-CoA dehydrogenase (MCAD) deficiency; Short chain acyl-CoA dehydrogenase (SCHAD) deficiency; Systemic primary carnitine deficiency; Very long chain acyl-CoA dehydrogenase (VLCAD) deficiency |
| IMPLICATEDGENES | ACAD8, ACAD9, ACADM, ACADS, ACADSB, ACADVL, CPT1A, CPT2, ETFA, ETFB, ETFDH, HADH, HADHA, HADHB, HMGCL, HMGCS2, HSD17B10, MLYCD, SLC22A5, SLC25A20, TAFAZZIN |
中鎖アシルCoA脱水素酵素欠損症(Medium chain acyl-CoA dehydrogenase (MCAD) deficiency)
中鎖アシルCoA脱水素酵素欠損症(MCAD欠損症)は、体内で特定の脂肪をエネルギーに変換する能力に影響を与える、まれな遺伝性の疾患である。この疾患では、脂肪をエネルギー源として利用するために必要な中鎖アシルCoA脱水素酵素が欠損しているため、特に絶食時に深刻な影響が現れる。通常、食事をしていない時間帯には、体内に蓄えられた脂肪が燃料として使われるが、MCAD欠損症の人は中鎖脂肪酸を適切に分解できない。その結果、未分解の脂肪酸が体内に蓄積し、エネルギー不足に陥ることになる。
MCAD欠損症の症状は、通常、乳児期や幼児期に初めて現れるが、まれに成人するまで診断されないこともある。主な症状として、嘔吐、極度の倦怠感(疲れやすさ)、そして低血糖(低血糖症)が挙げられる。これらの症状が進行すると、発作(けいれん)、呼吸困難、肝機能障害、脳障害、昏睡、さらには突然死につながることがある。特に乳幼児の場合、発症時に適切な医療対応がなければ、命に関わる危険性が高くなる。
MCAD欠損症の症状を引き起こす主な要因は、長時間の絶食や病気による食欲不振である。特に、ウイルス感染症にかかったときは、食事の摂取量が減ることでエネルギー不足になりやすくなる。MCAD欠損症の患者は脂肪を適切に利用できないため、わずかに食事を抜くだけでも代謝のバランスが崩れ、重篤な症状を引き起こすことがある。
この病気は、かつてライ症候群(Reye症候群)と誤診されることがあった。ライ症候群は、水ぼうそうやインフルエンザの回復期に発症することがある重篤な疾患で、アスピリンの使用との関連が指摘されている。MCAD欠損症とライ症候群は症状が似ているため、誤診の原因となることがあった。そのため、正しい診断を早期に行うことが重要である。
MCAD欠損症の原因は、「ACADM」遺伝子の変異によるものである。この遺伝子は中鎖アシルCoA脱水素酵素の生成を担っているが、変異があると十分に機能する酵素が作られない。本疾患は常染色体劣性遺伝という遺伝形式をとる。これは、両親からそれぞれ変異した遺伝子を受け継いだ場合にのみ発症することを意味する。一方、変異遺伝子を1つだけ持つ保因者(キャリア)は通常、症状を示さないが、子どもに遺伝する可能性がある。
生化学的に見ると、MCAD欠損症はミトコンドリア脂肪酸β酸化異常症に分類される。これは、体が脂肪をエネルギーに変換するためのβ酸化という代謝経路に障害があることを意味する。MCAD欠損症の患者には、以下のような代謝異常が見られる。低ケトン性低血糖(通常、低血糖時にはケトン体が増えるはずだが、MCAD欠損症ではケトン体が不足する)、中鎖ジカルボン酸尿症(未分解の脂肪酸が尿中に排出される)、血液や組織のカルニチン不足(脂肪酸をミトコンドリアに運ぶために必要な物質が不足する)などである。これらの異常が重なることで、MCAD欠損症の患者は代謝が不安定になり、エネルギー不足に陥りやすくなる。
MCAD欠損症は重篤な疾患であるが、早期診断と適切な管理により、予後を大幅に改善できる。現在、多くの国では新生児スクリーニング検査の一環としてMCAD欠損症の検査が行われており、これにより早期発見と適切な対応が可能になっている。
管理の基本は、絶食を避けることである。食事の間隔を一定に保ち、長時間の絶食を防ぐことが推奨される。また、病気や体調不良の際には、低血糖や代謝異常を防ぐために適切な治療を受けることが必要である。さらに、一部の患者にはカルニチン補充が推奨されることがある。カルニチンは、脂肪酸をミトコンドリアに運び、エネルギーとして利用するのを助ける重要な物質である。
適切な管理を行うことで、MCAD欠損症の患者も健康的な生活を送ることが可能である。しかし、食事管理が不十分だったり、病気のリスクについて十分な理解がなかったりすると、重篤な合併症を引き起こす可能性がある。そのため、この病気について正しく理解し、早期に症状を認識し、予防策を徹底することが、MCAD欠損症の管理には不可欠である。
短鎖アシルCoA脱水素酵素欠損症(Short chain acyl-CoA dehydrogenase (SCHAD) deficiency)
短鎖アシルCoA脱水素酵素欠損症(SCAD欠損症)は、まれな遺伝性の代謝異常であり、特定の脂肪をエネルギーに変換する能力が低下する病気です。特に、絶食時や病気の際に影響が現れやすく、体が十分なエネルギーを作り出せなくなります。この病気は、脂肪酸酸化異常症(Fatty Acid Oxidation Disorders, FODs)と呼ばれる代謝疾患の一種で、脂肪をエネルギー源として利用する機能に障害が生じるのが特徴です。
SCAD欠損症の原因は、ACADS遺伝子の変異にあります。この遺伝子が正常に働かないと、短鎖アシルCoA脱水素酵素(short-chain acyl-CoA dehydrogenase)の量が不足します。この酵素は、短鎖脂肪酸を分解し、エネルギーに変換する過程で重要な役割を果たします。しかし、この酵素が不足すると、脂肪酸やその他の代謝産物が体内に蓄積し、さまざまな症状を引き起こす可能性があります。
SCAD欠損症の症状や発症の仕方は非常に個人差が大きく、同じ家族内でも異なる症状を示すことがあります。一部の人はまったく症状がなく、新生児スクリーニングや遺伝子検査で偶然発見されることもあります。一方で、幼少期や成人になってから症状が現れる場合もあります。乳幼児期に発症する場合、哺乳不良(ミルクや食事をうまく摂れない)、嘔吐、低血糖(血糖値が下がることで意識がもうろうとすることもある)、嗜眠(元気がなく、眠気が強い状態)、発育不良(成長が遅れる)、筋力の低下(体がふにゃふにゃして力が入りにくい)、発達の遅れ、けいれん発作などの症状がみられることがあります。また、一部の乳児では、頭囲が通常より小さくなる小頭症(microcephaly)が見られることもあります。これらの症状は、絶食や病気、その他の代謝ストレスによって悪化することがあります。
一方で、成人期になってから発症するケースもあります。その場合、筋力低下や進行性の筋萎縮症(myopathy)として現れることが多いです。しかし、SCAD欠損症の遺伝子変異を持っていても、一生涯にわたって症状が現れない人もいます。新生児スクリーニングの普及により、SCAD欠損症に関連する代謝の異常を持っていながらも健康に過ごしている人が多く発見されるようになりました。そのため、専門家の間ではSCAD欠損症を「特定の疾患」ではなく、「生化学的な特徴の一つ」として捉える考え方が広まっています。現在も、SCAD欠損症がどの程度医学的に重要な意味を持つのかについては議論が続いています。
SCAD欠損症の症状は、ライ症候群(Reye syndrome)などの他の代謝異常と似ていることがあります。ライ症候群は、ウイルス感染から回復しつつある子どもに発症することがある重篤な病気で、特にアスピリンの使用によって悪化することが知られています。SCAD欠損症と症状が重なることがあるため、正確な診断が非常に重要になります。
SCAD欠損症は、常染色体劣性(autosomal recessive)の遺伝形式で受け継がれます。このため、発症するには、両親それぞれから変異したACADS遺伝子を1つずつ受け継ぐ必要があります。一方で、片方の遺伝子だけが変異している保因者(キャリア)は、通常、症状を示しません。
SCAD欠損症の重症度には個人差が大きいのも特徴の一つです。ある人は、代謝危機(metabolic crisis)と呼ばれる深刻な状態に陥ることがあります。一方で、生涯を通じて健康に過ごす人もいます。同じ遺伝子変異を持っていても、症状の有無や重症度が異なることから、遺伝的要因だけでなく、環境要因や他の遺伝的要素も影響していると考えられています。このような背景から、SCAD欠損症は単一の疾患ではなく、「スペクトラム(幅のある)疾患」として理解されることが増えています。
SCAD欠損症の治療方法は、症状の有無や重症度によって異なります。多くの人は特別な治療を必要としません。しかし、症状がある場合は、絶食を避けること、バランスの取れた食事を維持すること、代謝ストレスの兆候を注意深く観察することが推奨されます。場合によっては、脂肪酸の代謝を助けるカルニチンや、酵素の働きを助けるリボフラビン(ビタミンB2)などの栄養補助食品が処方されることもあります。
SCAD欠損症は、症状の予測が難しく、無症状のケースも多いため、現在も研究が進められている分野です。なぜ一部の人には症状が出るのに、他の人には出ないのか、症状がある場合、どのように管理するのが最適かといった点について、今後もさらなる研究が進められることが期待されています。
全身性一次性カルニチン欠損症(Systemic primary carnitine deficiency)
全身性カルニチン欠乏症(CDSP)は、体が脂肪をエネルギーとして適切に利用できなくなる、まれな遺伝性疾患です。この病気は、高親和性カルニチントランスポーター(OCTN2)と呼ばれるタンパク質の異常によって引き起こされます。OCTN2は、細胞内にカルニチンを取り込み、体外への過剰な排出を防ぐ働きを持っています。しかし、SLC22A5遺伝子の変異によってこのトランスポーターの機能が低下すると、脂肪酸を適切に酸化できなくなります。脂肪酸の酸化は、特に空腹時や病気の際に重要なエネルギー産生のプロセスであるため、この機能が損なわれるとさまざまな症状が現れます。CDSPは常染色体劣性遺伝の形式で受け継がれるため、発症には両親からそれぞれ変異した遺伝子を受け継ぐ必要があります。
カルニチンは、主に食事を通じて摂取される自然由来の物質で、ミトコンドリア(細胞内でエネルギーを生み出す器官)へ脂肪酸を運ぶ重要な役割を担っています。しかし、CDSPの患者では、カルニチンが適切に吸収・保持されないため、血液や組織内のカルニチン濃度が著しく低下します。その結果、特にエネルギー消費が激しい心臓、筋肉、肝臓といった臓器で、エネルギーを効率的に産生できなくなります。
CDSPの症状は個人によって大きく異なります。無症状のまま過ごせる人もいれば、深刻な健康問題を抱える人もいます。 多くの場合、乳児期や幼児期に発症しますが、成人になってから症状が現れるケースもあります。主な症状には、拡張型心筋症(心臓の拡大と機能低下)、筋力低下、低血糖(血糖値の異常低下)、肝機能障害、嘔吐、意識障害、脳症(脳の機能障害) などがあります。重症の場合、成長不良や再発性のけいれん発作が見られることもあり、空腹時や感染症が発症の引き金となることが少なくありません。さらに、適切な治療を受けない場合、突然の心不全を引き起こし、命に関わることもあります。
この疾患の大きな特徴の一つは、早期診断と適切な治療により、症状が完全に回復する可能性があることです。カルニチンを補充することで血液中のカルニチン濃度を正常化し、脂肪の代謝が適切に行われるようになります。これにより、ほとんどの患者の症状が完全に改善されます。 しかし、診断や治療が遅れると、心不全や重篤な神経障害へと進行するリスクが高まります。
CDSPは症状の多様性のため、他の代謝疾患と誤診されることがあります。たとえば、ライ症候群(Reye症候群)は、インフルエンザや水痘(水ぼうそう)などのウイルス感染症から回復途中の子どもに発症する重篤な疾患で、CDSPと似た症状を示すことがあります。そのため、CDSPが見逃され、大人になってから運動耐性の低下、慢性的な疲労、筋力低下といった軽度の症状のみが現れることもあります。
CDSPは、心臓、骨格筋(筋肉)、腎臓、肝臓などの複数の臓器に影響を及ぼします。特に腎臓では、カルニチンが適切に再吸収されず、尿中に過剰に排泄される(腎性カルニチン喪失)ことが、カルニチン欠乏をさらに悪化させます。これにより、エネルギー産生だけでなく、ケトン体の生成(肝臓がエネルギー源としてケトン体を作るプロセス)も妨げられます。そのため、CDSPの患者は、低血糖であるにもかかわらず、ケトン体が十分に生成されない「低ケトン性低血糖」を起こしやすくなり、代謝バランスが不安定になりやすいのです。
この疾患は深刻な健康リスクを伴う可能性があるものの、早期に適切な治療を受ければ完全に管理可能な数少ない代謝疾患の一つです。近年、一部の地域では新生児スクリーニング検査が実施されており、これにより症状が現れる前に診断を受け、カルニチン補充を開始できるケースが増えています。すでに症状が出ている場合でも、適切な治療を受けることで生活の質を大幅に向上させ、長期的な合併症を防ぐことができます。
総じて、全身性カルニチン欠乏症は、カルニチンの輸送異常によって引き起こされるまれな代謝疾患です。この病気により、体が脂肪をエネルギーとして適切に利用できなくなり、軽い疲労感から生命を脅かす臓器障害まで、さまざまな症状が現れる可能性があります。しかし、早期診断とカルニチン補充療法により、ほぼすべての症状が予防・改善可能であり、適切な管理を行えば良好な予後が期待できます。
極長鎖アシルCoA脱水素酵素欠損症(Very long chain acyl-CoA dehydrogenase (VLCAD) deficiency)
超長鎖アシルCoA脱水素酵素(VLCAD)欠損症は、体内で特定の脂肪をエネルギーに変換する能力が低下するまれな遺伝性疾患です。特に、ミトコンドリア内での超長鎖脂肪酸の酸化が正常に行われなくなります。この酸化は、空腹時にエネルギーを生み出すために不可欠なプロセスであり、これが機能しないと、体は蓄えられた脂肪を適切にエネルギーに変換できなくなります。その結果、さまざまな症状や合併症が引き起こされる可能性があります。
VLCAD欠損症は、ACADVL遺伝子の変異によって発症し、常染色体劣性遺伝の形式で受け継がれます。つまり、発症には両親からそれぞれ変異した遺伝子を受け継ぐ必要があります。この疾患は、ミトコンドリア脂肪酸β酸化異常の一つであり、他にも中鎖アシルCoA脱水素酵素(MCADD)欠損症や短鎖アシルCoA脱水素酵素(SCADD)欠損症などの関連疾患が存在します。かつて「長鎖アシルCoA脱水素酵素(LCAD)欠損症」と診断されていた患者の多くが、後にVLCAD欠損症であることが明らかになりました。
VLCAD欠損症の症状や発症年齢には個人差が大きく、一般的に3つのタイプに分類されます。最も重症な早期発症型は、生後間もなく症状が現れ、心筋症(心臓の筋肉の障害)やケトン体を伴わない低血糖(低血糖性ケトーシス不全)といった生命を脅かす合併症を引き起こし、死亡率も高いとされています。次に、中間型は、幼児期から小児期にかけて発症し、繰り返し低血糖を引き起こすものの、早期発見と適切な管理によって比較的予後が良いとされています。最後に、成人発症型は主に筋肉に関連する症状が特徴です。運動後に筋肉痛や筋力低下を感じることが多く、横紋筋融解症(筋肉の細胞が破壊される状態)を引き起こすことがあります。これにより、筋肉タンパク質(ミオグロビン)が尿中に排泄される(ミオグロビン尿症)ことがあり、腎臓に負担をかける可能性もあります。症状は、長時間の絶食や激しい運動、発熱を伴う病気などが引き金となることが多いです。
VLCAD欠損症の主な症状には、低血糖、エネルギー不足、筋力低下が挙げられます。重症の場合、肝臓や心臓の機能に影響を及ぼすこともあります。特に乳児や幼児では、病気や空腹時にエネルギーの需要が増加するため、重篤な代謝危機を引き起こすリスクが高くなります。
この疾患の管理には、食事療法が重要となります。具体的には、脂肪の利用を抑え、炭水化物を多く含む食事を摂ることが推奨されます。また、中鎖脂肪酸(MCT:ミディアムチェーントリグリセリド)を補給することで、体が代替エネルギー源として利用できるようにする方法も用いられます。特に、絶食を避け、エネルギー摂取を適切に管理することが不可欠です。心臓に深刻な影響がある場合は、追加の医療的介入が必要になることもあります。
VLCAD欠損症は、多くの国で新生児スクリーニング(生後すぐに行われる代謝異常の検査)の対象となっており、早期発見が可能です。早期診断によって適切な食事管理や治療を開始することで、重篤な合併症を防ぎ、予後を大きく改善できる可能性があります。
この疾患は、脂肪酸の代謝がエネルギー産生においていかに重要であるかを示す代表例です。また、遺伝子検査の意義や、早期介入の重要性を改めて浮き彫りにしています。VLCAD欠損症に関する理解が深まり、適切な管理が進むことで、より多くの患者の生活の質を向上させることが期待されています。
グリシン脳症(Glycine encephalopathy)
| REPRESENTATIVEDISEASES (JA) | グリシン脳症、高グリシン血症(乳酸アシドーシスおよび発作) |
|---|---|
| REPRESENTATIVEDISEASES (EN) | Glycine encephalopathy; Hyperglycinemia (Lactic acidosis and seizures) |
| IMPLICATEDGENES | AMT, GCSH, GLDC, LIAS, SLC6A9 |
グリシン脳症(Glycine encephalopathy)
グリシン脳症(Glycine Encephalopathy, GE)、または非ケトン性高グリシン血症(Nonketotic Hyperglycinemia, NKH)は、グリシンの代謝異常によって引き起こされる非常にまれな遺伝性の代謝疾患です。この病気では、グリシンというアミノ酸が体内、とくに脳内に異常に蓄積し、それに伴い重篤な神経症状が現れます。遺伝形式は常染色体劣性遺伝であり、発症するにはAMT、GLDC、GCSHといった遺伝子に変異を持つ両親から、それぞれ変異した遺伝子を受け継ぐ必要があります。これらの遺伝子に異常があると、グリシンを分解する酵素が正常に機能せず、結果としてグリシンが体内に蓄積してしまいます。
この病気にはいくつかのタイプがあり、主に症状の重さや発達の進行度によって分類されます。最も一般的で重症なタイプは新生児型グリシン脳症(classical neonatal glycine encephalopathy)で、生後数日以内に症状が現れます。影響を受けた新生児は、極度の眠気(傾眠)、哺乳困難、筋力の低下(筋緊張低下)、呼吸障害、けいれんなどを発症することがあります。適切な治療を行わない場合、症状は急速に悪化し、昏睡状態に至ることもあります。初期の危機を乗り越えたとしても、多くの子どもは著しい発達の遅れ、知的障害、制御が困難なてんかんを抱えることになります。ほとんどの場合、座る、物をつかむ、話すといった基本的な発達のマイルストーンを達成することができず、仮に一時的に習得できたとしても、後に失われることがあります。
一方で、軽症型グリシン脳症(attenuated glycine encephalopathy)では、比較的軽度の症状がみられます。発達の遅れはあるものの、時間をかけて歩行やコミュニケーション(手話を含む)を習得できる場合があります。また、けいれん発作が起こることもありますが、重症型と比べると治療による管理が比較的容易です。その他、筋緊張の異常(筋緊張亢進)、意図しない不規則な動きを引き起こす舞踏病(chorea)、多動などの症状がみられることもあります。MRI(磁気共鳴画像診断)を行うと、重症型の患者では脳の左右をつなぐ脳梁(のうりょう、corpus callosum)が通常よりも小さいといった異常がみられることがあります。
また、まれなケースとして一過性グリシン脳症(transient glycine encephalopathy)というタイプもあります。このタイプでは、新生児型と似た症状が現れますが、時間の経過とともにグリシンの蓄積が減少し、症状が改善するのが特徴です。完全に健康になるわけではありませんが、重症型と比較すると予後(病気の経過や治療後の見通し)は良好で、発達の進行が期待できます。
さらに、ごくまれなケースとして孤発性非ケトン性高グリシン血症(isolated nonketotic hyperglycinemia)があります。このタイプでは、新生児期に昏睡、重度の筋緊張低下、ミオクローヌス発作(突然の筋肉のけいれん)、小頭症(頭の大きさが通常よりも小さい状態)などの深刻な神経症状がみられます。このタイプの多くは、重度の知的障害や運動障害を伴います。
グリシン脳症の診断には、臨床症状、生化学的検査、遺伝子検査を組み合わせた総合的な評価が必要です。血液や脳脊髄液(cerebrospinal fluid, CSF)のグリシン濃度が高いことが、診断の重要な手がかりとなります。また、追加の検査によって、グリシン代謝に関わる酵素の活性低下を確認することも可能です。さらに、遺伝子検査を行うことで、原因となる特定の遺伝子変異を特定できます。
現在のところ、グリシン脳症を根本的に治療する方法は存在しません。治療は主に症状の管理を目的としており、抗てんかん薬やグリシン濃度を下げるための治療が行われます。一部の患者では、食事療法や神経伝達物質のバランスを調整する薬が有効な場合もありますが、病気の重症度によって治療の効果は大きく異なります。最も重症なケースでは寿命が短縮されることが多い一方で、軽症型の患者は適切なケアを受けることで、ある程度の自立した生活が可能になることもあります。
グリシン脳症は、重症度によって症状の幅が広い疾患ですが、いずれのタイプであっても患者やその家族にとって大きな負担となる深刻な病気です。近年、医学の進歩によって診断の精度や症状の管理は改善されてきていますが、今後の研究によってより効果的な治療法が開発され、長期的な予後が向上することが期待されています。
高グリシン血症(Hyperglycinemia (Lactic acidosis and seizures))
高グリシン血症・乳酸アシドーシス・けいれん(HGCLAS)は、非常にまれで重篤な遺伝性疾患であり、生後すぐに発症します。この病気は、両親からそれぞれ1つずつ異常な遺伝子を受け継ぐことで発症する常染色体劣性遺伝の形式をとります。主な特徴として、筋肉の緊張が低下する筋低緊張や、けいれん発作が現れることに加え、血液中のグリシンと乳酸の濃度が上昇します。これらの異常によって深刻な神経障害が引き起こされ、脳の機能が大きく低下する脳症や、運動や精神の発達が遅れる発達遅滞が見られます。多くの場合、幼少期のうちに病状が進行し、命に関わる状態になることもあります。
HGCLASは、変異型非ケトン性高グリシン血症(nonketotic hyperglycinemia, NKH)と呼ばれる病態群の一つに分類されます。通常の(古典的な)NKHでは、脳脊髄液(CSF)中のグリシン濃度が極端に上昇することが特徴ですが、HGCLASでは血漿や尿中のグリシン濃度が上昇し、それに加えて深刻な代謝異常を伴います。研究によると、HGCLASはミトコンドリアにおけるリポ酸の生合成異常によって引き起こされると考えられています。リポ酸は、細胞がエネルギーを作り出す代謝に必要な物質ですが、その合成に異常があると、細胞内の重要な代謝経路が正常に機能しなくなります。特に、ピルビン酸の酸化が障害されることで、エネルギーを生み出すプロセスが滞り、結果として体内に乳酸が異常に蓄積する乳酸アシドーシスが発症します。この乳酸アシドーシスは進行性の神経障害を引き起こし、病状の悪化につながります。
臨床的には、HGCLASは重度の脳筋症(脳と筋肉の両方に影響を及ぼす疾患)として現れ、深刻な発達遅滞や成長不良を引き起こします。多くの患者は新生児期からてんかん発作を伴い、適切な栄養を摂取しても十分に成長できないことが多く、また筋低緊張が持続するのも特徴的です。乳酸アシドーシスや代謝異常、さらには神経症状が複雑に絡み合うため、治療が非常に困難であり、現在のところ根本的な治療法は確立されていません。
HGCLASは、その重症度や進行の速さから、代謝疾患やミトコンドリア病の研究分野において重要な課題とされています。早期診断を行い、適切な支持療法を提供することが重要ですが、現時点では長期的な予後は依然として厳しい状況です。今後の研究が進むことで、より効果的な治療法の開発が期待されています。
グリコーゲン貯蔵疾患(Glycogen storage diseases)
| REPRESENTATIVEDISEASES (JA) | グリコーゲン貯蔵疾患 Ia(フォン・ギールケ病); グリコーゲン貯蔵疾患 II(ポンペ病) |
|---|---|
| REPRESENTATIVEDISEASES (EN) | Glycogen storage disease Ia (Von Gierke disease); Glycogen storage disease II (Pompe disease) |
| IMPLICATEDGENES | AGL, ALDOA, ALDOB, CPT2, ENO3, FBP1, G6PC, GAA, GBE1, GYG1, GYS1, GYS2, LAMP2, LDHA, PFKM, PGAM2, PHKA1, PHKA2, PHKB, PHKG2, PRKAG2, PYGL, PYGM, SLC2A2, SLC37A4 |
グリコーゲン貯蔵疾患 Ia(フォン・ギールケ病)(Glycogen storage disease Ia (Von Gierke disease))
グリコーゲン貯蔵病Ia型(GSDIa、フォンク・ギールケ病とも呼ばれる)は、非常にまれな遺伝性の代謝疾患です。この病気は、グルコース-6-ホスファターゼ(G6Pase)という酵素が欠損することで発症します。この酵素は、グリコーゲン分解(グリコーゲノリシス)や糖新生(グルコネオジェネシス)といった代謝経路の最終段階で重要な役割を果たし、体内でブドウ糖を作り出し血糖値を維持するために不可欠です。しかし、G6Paseが正常に機能しないと、血液中にブドウ糖を供給できず、肝臓や腎臓にグリコーゲンや脂肪が過剰に蓄積し、深刻な代謝異常を引き起こします。
この疾患の症状は通常、生後数か月で現れ、特に生後三〜四か月頃に顕著になります。この時期になると赤ちゃんは夜間に長く眠るようになり、授乳の間隔が長くなりますが、GSDIaの子どもは血糖値を一定に保つことができず、低血糖(重度の血糖低下)を起こしやすくなります。低血糖が進行すると、けいれんや極度の倦怠感を引き起こすことがあります。さらに、乳酸アシドーシス(血液中の乳酸が過剰に蓄積する状態)、高脂血症(血液中の脂肪が異常に増える状態)、高尿酸血症(尿酸値が異常に高くなる状態)といった代謝異常を伴うことが多く、これらが痛風や膵炎などのさらなる合併症につながることもあります。
GSDIaの子どもは、肝臓にグリコーゲンが蓄積することで肝腫大(肝臓の肥大)を引き起こします。そのため、お腹が膨らんで見えることが多く、腎臓も大きくなることがあります。また、この病気の影響で成長が遅れやすく、低身長や手足の細さが特徴的です。さらに、思春期の発達が遅れたり、成長全般が遅れる傾向があります。成人に近づくにつれ、骨粗しょう症(骨がもろくなり、骨折しやすくなる状態)を発症することもあります。
GSDIaの長期的なリスクの一つに、肝腺腫(肝臓にできる良性の腫瘍)の発生があります。これらの腫瘍は通常良性ですが、一部は悪性化(肝がんへの進行)することがあります。また、その他の合併症として腎疾患や高血圧があり、女性では多嚢胞性卵巣症候群(ポリシスティック・オーバリー・シンドローム、PCOS)がみられることもあります。
この病気の原因は、G6PC遺伝子(十七番染色体上の17q21領域)の変異です。この遺伝子はG6Paseを作るために必要な情報を持っていますが、変異があるとG6Paseが正常に機能せず、ブドウ糖を血中に放出できなくなります。その結果、GSDIa特有の低血糖や代謝異常が発生します。
GSDIaと似ていますが異なる特徴を持つ病気に、グリコーゲン貯蔵病Ib型(GSDIb)があります。GSDIbは、細胞内でグルコース-6-リン酸(グルコース-6-ホスフェート)の輸送に関わる異常によって発症します。この疾患では、GSDIaと同じ代謝異常に加えて、好中球減少症(白血球の一種である好中球の不足)が見られ、細菌感染を繰り返しやすくなります。好中球減少症は一歳頃までに明らかになることが多く、GSDIbの患者の多くは、炎症性腸疾患(IBD)を発症しやすい傾向があります。また、歯や口腔の異常(虫歯、歯肉炎、歯周病、口内炎など)もよく見られます。これらの症状はGSDIb特有のものであり、GSDIaの患者には通常見られません。
GSDIaは深刻な合併症を引き起こす可能性がありますが、近年の治療法の進歩により、患者の予後は大きく改善されています。治療の中心となるのは厳密な食事管理です。血糖値を安定させるため、頻繁な炭水化物の摂取や、場合によっては夜間の持続的な栄養補給が必要になります。適切に管理すれば、GSDIaの患者でも正常な成長や思春期の発達を迎え、多くの場合、成人まで健康的に生活することが可能です。
現在、GSDIaの根本的な治療法として、酵素補充療法や遺伝子治療の研究が進められています。これらの治療法が実用化されれば、病気の根本的な改善が期待され、患者の生活の質がさらに向上する可能性があります。
グリコーゲン貯蔵疾患 II(ポンペ病)(Glycogen storage disease II (Pompe disease))
ポンペ病は、グリコーゲン蓄積症II型(GSD2)や酸性マルターゼ欠損症とも呼ばれる、非常に稀な遺伝性疾患です。この病気は、酸性α-グルコシダーゼ(GAA)という酵素を作る遺伝子に変異が生じることで発症します。GAAは、細胞内のリソソームという構造内で、エネルギー源となるグリコーゲンを分解する重要な役割を担っています。しかし、この酵素の働きが不十分だと、リソソーム内にグリコーゲンが異常に蓄積し、特に筋肉細胞が深刻な影響を受けます。その結果、筋肉の機能が徐々に低下していきます。
ポンペ病の症状は非常に幅広く、発症する年齢や重症度は人によって大きく異なります。一般的に、病気の進行の仕方に応じて「古典的乳児発症型」「非古典的乳児発症型」「遅発型」の3つのタイプに分類されます。
最も重症な「古典的乳児発症型」は、生後数か月以内に発症します。このタイプの赤ちゃんは、著しい筋力低下(筋症)や筋肉の緊張の低下(低緊張)が見られるほか、肝臓の腫れ(肝腫大)や心臓の異常が特徴的です。特に、心臓が異常に大きくなる「心肥大」や、心筋の機能が低下する「心筋症」が起こり、生命を脅かす心疾患へと進行します。また、哺乳が難しくなることが多く、体重が増えず成長が遅れる(発育不良)といった問題も生じます。さらに、呼吸機能の低下による呼吸不全が深刻な合併症として現れます。このタイプは、適切な治療を受けなければ生後1年以内に心不全で命を落とすことがほとんどです。
「非古典的乳児発症型」は、古典的乳児発症型よりも発症がやや遅く、生後1年以内に症状が現れるのが一般的です。このタイプも重症ですが、病気の進行は比較的ゆっくりしています。運動機能の発達が遅れ、寝返りやお座りが難しくなるほか、筋力の低下が徐々に進行します。心肥大が見られることもありますが、心不全に至る可能性は古典的乳児発症型よりも低いとされています。ただし、呼吸機能の低下は深刻で、治療が行われなければ幼児期の早い段階で命を落とすことが多いです。
「遅発型ポンペ病」は、乳児期以降、幼児期・思春期・成人期と、さまざまな時期に発症する可能性があります。乳児発症型と比べると症状は軽いことが多く、心疾患を伴うことはほとんどありません。しかし、骨格筋、特に脚や胴体の筋肉が徐々に衰え、動作が困難になっていきます。また、呼吸を助ける筋肉も影響を受けるため、長期的には呼吸不全を引き起こすことがあります。病気の進行速度は個人差があり、発症後長い間ほとんど症状が現れないケースもあれば、徐々に運動障害や呼吸不全が深刻化するケースもあります。
ポンペ病のすべてのタイプに共通する原因は、酸性α-グルコシダーゼの欠損によるリソソーム内のグリコーゲン蓄積です。この異常な蓄積が細胞の働きを妨げ、特に筋肉組織において時間とともに衰弱や変性を引き起こします。
ポンペ病の予後(病気の進行や寿命)は、酵素の欠損の程度や臓器への影響の範囲によって大きく異なります。最も重症な古典的乳児発症型では、治療を受けなければほぼ確実に早期死亡につながります。一方で、遅発型では進行が緩やかですが、移動能力の低下や呼吸機能の障害など、日常生活に大きな支障をきたすことがあります。
ポンペ病は、「リソソーム病」と呼ばれる疾患の一種です。リソソーム病とは、リソソーム内の酵素が欠損することで、通常は分解されるはずの物質が蓄積し、細胞の働きを阻害する代謝疾患の総称です。ポンペ病の治療法としては、「酵素補充療法(ERT)」が大きく進歩しています。この治療では、欠損している酸性α-グルコシダーゼを人工的に補い、リソソーム内のグリコーゲンの蓄積を防ぎます。特に乳児発症型の患者の生存率向上に大きく貢献しており、多くの患者にとって画期的な治療法となっています。しかし、酵素補充療法だけでは病気の進行を完全に止めることはできません。そのため、早期診断と継続的な管理が非常に重要になります。
高インスリン血症性低血糖(Hyperinsulinemic hypoglycemia)
| REPRESENTATIVEDISEASES (JA) | 家族性高インスリン血症性低血糖症、タイプ 1、2、3、4、5、7、高インスリン血症-高アンモニア血症症候群 |
|---|---|
| REPRESENTATIVEDISEASES (EN) | Hyperinsulinemic hypoglycemia, familial, types 1, 2, 3, 4, 5, 7; Hyperinsulinism-Hyperammonemia syndrome |
| IMPLICATEDGENES | ABCC8, GCK, GLUD1, HADH, INSR, KCNJ11, SLC16A1 |
家族性高インスリン血症性低血糖症、タイプ1(Hyperinsulinemic hypoglycemia, familial, type 1)
高インスリン性低血糖症、家族性1型(Hyperinsulinemic Hypoglycemia, Familial, 1:HHF1)は、家族性高インスリン血症や先天性高インスリン血症とも呼ばれるまれな疾患です。この病気では、乳児期に膵臓から過剰なインスリンが分泌されることで、低血糖(血糖値の異常な低下)が持続的に起こります。通常、血糖値が下がるとインスリンの分泌も抑制されますが、HHF1ではこの調節機能が適切に働かず、血糖値が十分に低い状態でもインスリンが分泌され続けてしまいます。その結果、血糖値が危険なほど低下し、さまざまな健康リスクを引き起こす可能性があります。
この疾患は、症状の現れ方や重症度が患者ごとに異なることが特徴で、遺伝的にも多様性があります。遺伝の形式には、常染色体優性(片方の親から遺伝するタイプ)と常染色体劣性(両親の双方から遺伝するタイプ)の2種類が存在します。HHF1の一部の症例では、第11染色体にあるABCC8遺伝子に変異が確認されています。この遺伝子はインスリンの分泌調節に関与しており、変異があると通常の仕組みがうまく機能しなくなることがあります。特にABCC8遺伝子の変異によるHHF1は、一般的な治療薬であるジアゾキシド(インスリン分泌を抑える薬)が効きにくいことが知られています。
HHF1が適切に治療されない場合、低血糖発作を繰り返し、神経系への深刻な影響を及ぼす可能性があります。特に、発達の遅れやけいれん、脳損傷といった合併症が生じることもあるため、早期の診断と積極的な管理が極めて重要です。治療方法は、病気の重症度や遺伝子変異の種類によって異なります。軽度の場合は、インスリンの分泌を調整する薬を用いた治療が効果を示すことがあります。しかし、薬による治療が十分な効果を示さない重症例では、膵臓の一部を外科的に切除する手術が必要になることもあります。これにより、インスリンの過剰分泌を抑え、低血糖の発作を防ぐことが可能になります。
HHF1はまれな疾患であるため、診断が遅れることも少なくありません。そのため、医療従事者の間でこの疾患に対する認識を高めることが重要です。特に、新生児の時期から授乳がうまくできない、機嫌が悪い、ぐったりしている、けいれんを起こすといった症状が見られる場合は、早期に適切な検査を受けることが重要です。迅速な診断と治療によって、低血糖の影響を最小限に抑え、長期的な予後を改善することが期待されます。
高インスリン血症・高アンモニア血症症候群(Hyperinsulinism-Hyperammonemia syndrome)
インスリン分泌亢進・高アンモニア血症症候群(Hyperinsulinism-Hyperammonemia syndrome、HI/HA)は、GLUD1遺伝子の変異によって引き起こされるまれな先天性疾患です。この遺伝子は染色体10q23.3に存在し、グルタミン酸脱水素酵素(GDH)という酵素の働きを調節する役割を担っています。通常、この酵素の活動はグアノシン三リン酸(GTP)によって抑えられていますが、GLUD1遺伝子の変異により調節機能が失われ、GDHの活性が異常に高まります。GDHは膵臓のランゲルハンス島、肝臓、腎臓、脳に存在し、それぞれの臓器でエネルギー代謝に関与しています。しかし、この酵素が過剰に活性化すると、異常に大量のインスリンが分泌されるとともに、血中のアンモニア濃度が上昇(高アンモニア血症)するという二つの主要な異常が引き起こされます。この疾患は、ジアゾキシド感受性高インスリン血症の代表的な一つであり、インスリン分泌を抑える薬剤であるジアゾキシドに比較的よく反応します。
HI/HAの原因となるGLUD1遺伝子の変異の約70%は自然発生的(de novo)に起こります。残りの30%は常染色体優性遺伝によるもので、親がこの疾患を持つ場合、子どもに遺伝する確率は50%となります。生まれつきこの疾患を持っていても、症状がはっきり現れるのは生後4〜12か月頃が多いとされています。
HI/HAの最も顕著な特徴は、異常なインスリン分泌による低血糖(低血糖症)の発作を繰り返すことです。一般的な高インスリン血症と異なり、HI/HAの患者は糖分よりもタンパク質(特にロイシン)に対して異常に強く反応し、インスリンを過剰に分泌することが分かっています。これはタンパク質誘発性低血糖と呼ばれ、ロイシンがGDHを活性化させることでさらにインスリンの分泌が促進されることが原因です。そのため、患者は高タンパク食を摂取した後に低血糖を発症することが多いですが、糖分摂取によるインスリンの過剰分泌は起こりません。
また、血中のアンモニア濃度が慢性的に高いにもかかわらず、HI/HAの患者は一般的な高アンモニア血症の症状である意識混濁や倦怠感、脳症などをほとんど示さないことが特徴です。この理由は明確には分かっていませんが、体が長期間のアンモニア上昇に適応している可能性が考えられています。しかし、長期的な影響として神経系への障害が懸念されており、てんかん、認知機能の低下、行動異常といった合併症が報告されています。HI/HAに関連するてんかんの正確な発症メカニズムはまだ解明されていませんが、脳内のGDHが過剰に活性化することで、神経の興奮を促すグルタミン酸と、神経の働きを抑制するGABAのバランスが崩れることが一因と考えられています。このバランスの乱れが発作の引き金となる可能性があります。
HI/HAの治療には、ジアゾキシドが主に用いられます。この薬はインスリンの分泌を抑制し、低血糖発作を防ぐ効果があります。しかし、治療を行わずにいると低血糖の発作が繰り返され、脳に深刻なダメージを与える可能性があります。その結果、知的発達の遅れや発達障害につながるリスクも高まるため、早期診断と適切な管理が非常に重要です。薬による治療が不十分な場合、食事療法(高タンパク食の制限)や、重症例では膵臓の一部切除などの外科的治療が検討されることもあります。
HI/HAは代謝に関わる複雑な疾患であり、低血糖や神経合併症を防ぐためには継続的な管理が必要です。しかし、適切な治療を行うことで、患者の生活の質を大きく向上させることが可能です。
高フェニルアラニン血症(Hyperphenylalaninemia)
| REPRESENTATIVEDISEASES (JA) | 高フェニルアラニン血症 A BH4欠乏症; フェニルアラニン水酸化酵素欠損症 |
|---|---|
| REPRESENTATIVEDISEASES (EN) | Hyperphenylalaninemia A BH4-Deficiency; Phenylalanine hydroxylase deficiency |
| IMPLICATEDGENES | DNAJC12, GCH1, PAH, PCBD1, PTS, QDPR |
高フェニルアラニン血症 A BH4欠乏症(Hyperphenylalaninemia A BH4-Deficiency)
テトラヒドロビオプテリン(BH4)欠乏による高フェニルアラニン血症(HPA)は、神経伝達物質の合成に影響を及ぼし、さまざまな神経症状を引き起こす希少な遺伝性疾患です。一般的なフェニルケトン尿症(PKU)はPAH遺伝子の変異によって生じ、フェニルアラニンの摂取制限による管理が可能ですが、BH4欠乏によるHPAは、BH4の合成や再生に関わる遺伝子の異常によって発症する常染色体劣性疾患です。
BH4は、フェニルアラニンヒドロキシラーゼ、チロシンヒドロキシラーゼ、トリプトファンヒドロキシラーゼといった酵素の働きを助ける補因子であり、フェニルアラニンの代謝やドーパミン・セロトニンなどの神経伝達物質の合成に必要不可欠です。そのため、BH4が欠乏すると、血中のフェニルアラニン濃度が上昇するだけでなく、ドーパミンやセロトニンの不足が生じ、認知機能や運動機能の低下につながります。
BH4欠乏によるHPAの中で最も一般的なタイプは、PTS遺伝子の変異によるHPABH4Aです。他にも、GCH1遺伝子の変異によるHPABH4B、QDPR遺伝子の変異によるHPABH4C、PCBD1遺伝子の変異によるHPABH4Dなどがあり、これらの疾患は全体の高フェニルアラニン血症患者の約1〜3%を占めるとされています。症状の現れ方には個人差があり、正常な発達を示す軽症例もあれば、筋緊張の低下(低緊張)、発達遅滞、ジストニアやジストニア・パーキンソニズムといった運動障害を伴う重症例もあります。
BH4欠乏症の影響は、特に運動機能や気分の調整、神経系の健康に重要なドーパミンやセロトニンの合成に及ぶため、深刻な神経症状を引き起こすことがあります。BH4欠乏によるHPAは、PKUのようにフェニルアラニン制限食のみでは改善されないため、別の治療法が必要です。治療には、BH4の補充や、ドーパミン・セロトニンの前駆体であるL-ドーパや5-ヒドロキシトリプトファンの投与が行われます。しかし、治療の効果は酵素の欠損の程度や治療開始の時期によって異なります。
また、BH4欠乏に関連する疾患の中には、必ずしも高フェニルアラニン血症を伴わないものもあります。例えば、SPR遺伝子の変異によるドーパ反応性ジストニアや、GCH1遺伝子の変異による常染色体優性ドーパ反応性ジストニア(DYT5)が挙げられます。これらの疾患の患者は、ストレスなどの要因によって一時的に高フェニルアラニン血症を発症することがあり、BH4の代謝と神経伝達物質の密接な関係を示しています。
現在、BH4欠乏症の理解を深め、より効果的な治療法を確立するために、患者データの収集や治療効果の検討が進められています。早期診断と適切な治療を行うことで、神経発達の促進や生活の質の向上が期待されます。BH4の代謝は複雑であり、さまざまな生化学的経路に影響を及ぼすため、今後も治療法の改善や新たな知見の蓄積が求められます。
フェニルアラニン水酸化酵素欠損症(Phenylalanine hydroxylase deficiency)
フェニルアラニン水酸化酵素(PAH)欠損症は、一般的にフェニルケトン尿症(PKU)と呼ばれる遺伝性の代謝異常です。この疾患では、体が必須アミノ酸であるフェニルアラニンを適切に代謝できず、血液中のフェニルアラニン濃度が異常に高くなります。その結果、軽度の高フェニルアラニン血症(HPA)から重症の古典的PKUまで、症状の程度はさまざまですが、治療を受けなければ認知機能や発達に深刻な影響を及ぼす可能性があります。
最も重症な古典的PKUでは、PAHの働きがほぼ完全に失われ、血中フェニルアラニン濃度が危険なレベルまで上昇します。これを放置すると、重度の知的障害が不可逆的に進行します。一方、軽度のPKUや非古典的HPAでは、血中フェニルアラニン濃度が1,200 μmol/L(20 mg/dL)未満であれば、知的発達への影響は比較的少ないとされています。
新生児スクリーニングの導入により、PAH欠損症の予後は大きく改善されました。生後すぐに血液検査(ガスリー法など)によってフェニルアラニンの上昇を検出し、早期診断と治療が可能になったことで、従来は避けられなかった深刻な神経学的合併症を予防できるようになりました。1960〜70年代にかけて新生児スクリーニングが広く普及し、PKUによる重篤な影響が大幅に減少しました。しかし、適切な治療を受けている患者でも注意力や認知機能に影響が残ることがあり、現在の治療法では完全に代謝バランスを回復するのは難しいと考えられています。
治療の基本は、フェニルアラニンを制限した低タンパク食と、フェニルアラニンを含まない特殊ミルク(医療用フォーミュラ)の併用です。古典的PKUの患者では、小児期の認知発達を最適化するために、血中フェニルアラニン濃度を120〜360 μmol/L(2〜6 mg/dL)に維持することが推奨されます。思春期以降の適切なフェニルアラニン目標値については明確な基準が定まっていませんが、一部の患者では補助療法が有効とされています。たとえば、サプロプテリン(BH4と呼ばれる補酵素の合成型)はPAHの活性を促進し、一部の患者のフェニルアラニン代謝を改善する可能性があります。また、大中性アミノ酸(LNAA)は脳内へのフェニルアラニンの取り込みを抑え、症状の改善に寄与すると考えられています。
PAH欠損症の適切な管理には、定期的な血液検査が不可欠です。血中フェニルアラニンやチロシンの濃度を測定しながら、成長や栄養状態をチェックし、発達の進捗状況を確認します。また、学習障害や情緒面の問題を早期に発見するために、心理学的な評価も推奨されます。
PKUの患者が特に避けるべきものとして、人工甘味料のアスパルテームが挙げられます。アスパルテームにはフェニルアラニンが含まれており、PKUの患者が摂取すると血中フェニルアラニン濃度が急激に上昇し、健康に悪影響を及ぼす可能性があります。
PAH欠損症の女性は、妊娠時に特別な注意が必要です。血中フェニルアラニン濃度が高い状態が続くと、胎児に深刻な影響を与えることが知られています。例えば、先天性心疾患、小頭症、知的障害などのリスクが高まり、この状態は母体PKU症候群(MPKU症候群)と呼ばれます。これを防ぐため、妊娠を考えている女性は、妊娠数か月前から厳格なフェニルアラニン制限食を実施し、妊娠中も頻繁に血液検査を行いながら、推奨される血中フェニルアラニン濃度を維持する必要があります。
PAH欠損症は常染色体劣性遺伝の形式をとります。これは、両親からそれぞれ変異したPAH遺伝子を1つずつ受け継ぐことで発症することを意味します。この遺伝子は12番染色体のq23.1領域に位置し、これまでに500種類以上の遺伝子変異が報告されています。遺伝子の型と病気の重症度の間には一定の関連がありますが、完全に一致するわけではありません。しかし、遺伝子検査を行うことで病気の重症度をある程度予測でき、リスクのある家族に対する遺伝カウンセリングにも役立ちます。
PAH欠損症の研究は、医学遺伝学の分野において重要な歴史を持っています。1934年にノルウェーの医師アスビョルン・フェリングによって初めて報告されたPKUは、世界で初めて新生児スクリーニングによって診断されるようになった遺伝性疾患です。20世紀半ばには、フェニルアラニンを制限する食事療法が有効であることが確認され、遺伝性疾患の管理における画期的な進展となりました。その後の早期介入により、患者の予後は大幅に改善されましたが、さらなる治療法の改善を目指した研究が現在も続いています。
しかし、現在の治療を受けていても、完全な代謝の安定化を達成することは難しく、多くの患者は注意力や実行機能に関する問題を抱えることがあります。また、思春期や成人後に食事管理を緩めると、可逆的または不可逆的な認知機能や情緒面の問題が生じる可能性があります。さらに、未診断のまま成人したPKU患者であっても、治療を開始し血中フェニルアラニン濃度を下げることで行動面の改善が見られることが報告されています。
遺伝子検査は、PAH欠損症の診断を確定し、家族内の保因者を特定する手段として利用できます。また、出生前診断の選択肢としても活用されます。PAH欠損症の患者の新生児の兄弟姉妹については、早期診断と治療のために生後すぐに血液検査を行うことが推奨されます。
PAH欠損症は健康上の課題を伴う疾患ではありますが、早期診断と厳格な食事管理、そして新しい治療法の活用により、多くの患者が健康的で充実した生活を送ることが可能です。しかし、長期的な合併症を最小限に抑えるためには、定期的な検査と継続的な治療の実施が不可欠です。
リソソーム蓄積障害(Lysosomal storage disorders)
| REPRESENTATIVEDISEASES (JA) | アスパルチルグルコサミン尿症、ファブリー病、ゴーシェ病(非定型)、SAP-B 欠乏による異染性白質ジストロフィー、ムコ多糖症 II 型(ハンター症候群)、ムコ多糖症 III 型(サンフィリッポ A、B、C、D)、ニーマン・ピック病 A、B、C、D 型、サンドホフ病、テイ・サックス病 |
|---|---|
| REPRESENTATIVEDISEASES (EN) | Aspartylglucosaminuria; Fabry disease; Gaucher disease (atypical); Metachromatic leukodystrophy due to SAP-B deficiency; Mucopolysaccharidosis type II (Hunter syndrome); Mucopolysaccharidosis Type III (Sanfilippo A, B, C and D), Niemann-Pick types A, B, C and D; Sandhoff disease; Tay-Sachs disease |
| IMPLICATEDGENES | AGA, ARSA, ARSB, ASPA, BTD, CLN3, CLN5, CLN6, CLN8, CTNS, CTSA, CTSK, DHCR7, FUCA1, GAA, GALC, GALNS, GBA, GCDH, GLA, GLB1, GM2A, GNE, GNPTAB, GNPTG, GNS, GUSB, HEXA, HEXB, HGSNAT, HPD, HRAS, HYAL1, IDUA, IDS, LAMP2, LIPA, MAN1B1, MAN2B1, MANBA, MCOLN1, MFSD8, NAGA, NAGLU, NEU1, NPC1, NPC2, PPT1, PSAP, SGSH, SLC17A5, SLC25A15, SMPD1, SUMF1, TPP1, VPS33 |
ムコ多糖症 II 型(ハンター症候群)(Mucopolysaccharidosis type II (Hunter syndrome))
ムコ多糖症Ⅱ型(MPS II)、別名ハンター症候群は、主に男性に発症するまれな遺伝性疾患です。この病気はX染色体に関連する劣性遺伝によって引き起こされ、体内のライソソーム酵素であるイズロン酸スルファターゼが不足または欠損することで発症します。この酵素は、グリコサミノグリカン(GAG)と呼ばれる糖鎖を分解する役割を持っていますが、酵素が十分に機能しないと、GAGが細胞内に蓄積し、全身の組織や臓器に影響を及ぼします。症状の現れ方や進行の度合いには個人差があり、知的発達の遅れや退行を伴う重症型と、知的発達が正常で症状が比較的軽い軽症型があります。
MPS IIの症状は、生まれた直後にはほとんど見られませんが、通常2歳から4歳頃に初めて現れます。初期の兆候として、特徴的な顔立ちの変化が見られ、鼻が広くなったり、唇が厚くなったり、頬がふくらんだりするほか、舌が大きくなる(巨舌症)こともあります。また、声帯が厚くなることで声が低くかすれたようになることがあります。さらに、気道が狭くなることで呼吸器感染症を繰り返しやすくなり、睡眠時無呼吸を引き起こすことも多く、医療的な処置が必要になる場合があります。成長は5歳頃までは正常に進みますが、それ以降は成長の速度が遅くなり、最終的には低身長となることが一般的です。また、関節が硬くなり変形することで、運動能力が制限されることもあります。
この疾患は全身のさまざまな器官や組織に影響を及ぼします。多くの患者で、通常より頭が大きくなる(大頭症)、脳内に過剰な液体がたまる(脳室拡大・水頭症)、肝臓や脾臓が肥大する(肝脾腫)といった症状が見られます。また、へそや鼠径部(そけいぶ)にヘルニアができやすいことも特徴です。皮膚は厚くなり、弾力が低下し、一部の患者では小さなこぶのような皮膚の隆起が見られます。さらに、聴力が低下することや、網膜の変性による視力障害が起こることもあります。手や指にしびれや脱力を引き起こす手根管症候群は小児期から発症しやすく、また、脊柱管が狭くなる脊柱管狭窄症により脊髄が圧迫されると、運動機能がさらに低下することがあります。心臓にも影響が及びやすく、多くの患者で心臓弁の異常が見られます。これが進行すると、心筋の肥大(心室肥大)、不整脈(脈の乱れ)が起こり、最終的に心不全に至ることもあります。
MPS IIは、大きく「神経型(重症型)」と「非神経型(軽症型)」の2つのタイプに分類されます。神経型では、幼少期から発達の遅れが見られ、その後、知的機能の低下が進行し、最終的には認知機能障害へとつながります。初期には多動や行動の問題が見られることが多いですが、病気が進行すると重度の知的障害や認知症のような症状が現れます。このタイプの患者は、呼吸障害や心疾患が原因で、20代から30代のうちに亡くなることが多いとされています。一方、非神経型の患者は知的発達が正常で、成人まで生存することが可能ですが、関節の変形や骨の異常(多発性骨異形成)、心疾患、呼吸器系の問題など、身体的な合併症を抱えることが少なくありません。
分子レベルでは、MPS IIの患者ではヘパラン硫酸やデルマタン硫酸といった糖鎖が細胞内のライソソームに蓄積し、その一部が尿中に排出されます。このような糖鎖の蓄積が細胞の正常な機能を妨げ、病気の進行につながると考えられています。従来、この疾患は「重症型」と「軽症型」に分類されていましたが、近年では病気の進行の違いをより適切に表すために「早期進行型」と「緩徐進行型」と呼ばれることもあります。
MPS IIは進行性の疾患であり、多くの合併症を伴いますが、医療の進歩により患者の生活の質は向上しています。症状の管理には、呼吸管理、理学療法、心臓の定期的な検査などが重要とされており、これらの適切なサポートを受けることで、より快適な生活を送ることが可能になります。
テイ・サックス病(Tay-Sachs disease)
テイ=サックス病は、主に神経系に影響を及ぼすまれな遺伝性の神経変性疾患です。この病気はHEXA遺伝子の変異によって引き起こされ、β-ヘキソサミニダーゼA(beta-hexosaminidase A)という酵素が十分に作られなくなります。この酵素は、GM2ガングリオシド(GM2 ganglioside)という脂質を分解する役割を持っていますが、機能しないと神経細胞(ニューロン)に蓄積し、特に脳や脊髄に深刻なダメージを与えます。これにより、神経細胞が徐々に破壊され、病状が進行していきます。テイ=サックス病は常染色体劣性遺伝(autosomal recessive inheritance)の形式で遺伝するため、両親ともにHEXA遺伝子に変異を持っている場合に発症します。世界のさまざまな集団に見られますが、特に東欧系アシュケナジム・ユダヤ人の間で発症率が高いことが知られています。
この病気の中で最も重症で一般的なのが乳児型(infantile Tay-Sachs disease)です。生後数か月までは通常通り成長しているように見えますが、生後3〜6か月頃から症状が現れ始めます。初期の兆候としては、筋力の低下や発達の遅れがあり、これまでに習得した寝返り、座る、はいはいといった動作ができなくなっていきます。また、大きな音に対して過剰に驚くという特徴的な反応が見られることもあります。病気が進行すると、けいれん発作、筋肉の不随意運動(ミオクローヌス)、飲み込みにくさ(嚥下障害)、視力や聴力の低下、知的機能の衰えといった症状が現れます。眼科検査では、「チェリー・レッド・スポット(cherry-red spot)」と呼ばれる特徴的な赤い斑点が確認されることが多いです。さらに病状が進むと、運動や嚥下の機能が失われ、意思疎通も困難になります。最良のケアを受けた場合でも、多くの乳児型患者は4歳を超えて生存することができません。
テイ=サックス病には、乳児型よりも発症時期が遅く、症状が比較的軽い若年型(juvenile form)や成人発症型(late-onset form)もあります。若年型は5歳頃から思春期の間に症状が現れ、筋力の低下、協調運動の困難(運動失調)、発話障害、認知機能の低下などが見られます。進行は乳児型ほど急激ではありませんが、多くの患者は10代のうちに合併症により命を落とします。
成人発症型は、比較的軽度で、症状の出方に個人差が大きいのが特徴です。初期症状としては、不器用さやぎこちない動作、筋力の徐々の低下、発話の問題、ふるえ(振戦)などがあり、進行するとうつ病や精神疾患(精神病など)を伴うこともあります。進行の速さも人によって異なり、一部の人は歩行補助が必要になるのに対し、通常に近い寿命を保つ人もいます。
テイ=サックス病の診断には、血液検査でβ-ヘキソサミニダーゼAの酵素活性を測定します。この酵素の活性がほぼゼロ、または極めて低い場合にテイ=サックス病が疑われます。また、HEXA遺伝子の遺伝子検査によって病気を確定診断することが可能です。この検査はキャリア(保因者)スクリーニングにも用いられ、特に発症リスクの高い集団では、保因者であるかどうかを事前に調べることが推奨されています。
現在のところ、テイ=サックス病に対する根本的な治療法は存在しません。治療は症状の管理と生活の質(QOL)の向上を目的としています。対症療法としては、けいれん発作の管理、呼吸補助、理学療法、栄養補助、嚥下が困難になった場合の経管栄養などが行われます。また、筋肉のこわばりや発作の症状を抑える薬物療法が用いられることもあります。成人発症型の患者では、精神面のサポートが必要となる場合もあります。
特に乳児型のテイ=サックス病は深刻な疾患ですが、近年の遺伝子スクリーニングや出生前診断の進歩により、発症リスクの高い集団における発症率は大幅に低下しています。現在、酵素補充療法、遺伝子治療、基質減少療法などの新しい治療法が研究されており、将来的により効果的な治療法が確立されることが期待されています。
メープルシロップ尿症とDLD欠乏症(Maple syrup urine disease and DLD deficiency)
| REPRESENTATIVEDISEASES (JA) | メープルシロップ尿症タイプIa、Ib、II、III |
|---|---|
| REPRESENTATIVEDISEASES (EN) | Maple syrup urine disease types Ia, Ib, II, III |
| IMPLICATEDGENES | BCKDHB, CKDHA, DBT, DLD, PPM1K |
メープルシロップ尿症タイプIa(Maple syrup urine disease type Ia)
メープルシロップ尿症(Maple Syrup Urine Disease, MSUD)は、非常にまれな遺伝性の代謝異常症で、体内で特定のアミノ酸を適切に分解できなくなる病気です。この病名は、患者の尿や耳垢が甘いメープルシロップのような独特のにおいを持つことに由来しています。MSUDは、ロイシン、イソロイシン、バリンといった分岐鎖アミノ酸を代謝する役割を担う「分岐鎖α-ケト酸脱水素酵素(branched-chain alpha-keto acid dehydrogenase, BCKAD)」の働きが低下または欠損することが原因で発症します。この酵素が正常に機能しないと、分岐鎖アミノ酸やその代謝産物が血液や組織に蓄積し、神経障害をはじめとする重篤な健康問題を引き起こします。
MSUDにはいくつかのタイプがあり、病気の重症度や治療への反応によって分類されます。最も一般的で重症度が高い「古典型MSUD」は、全体の約75%を占めています。このタイプでは、生後数日以内に哺乳がうまくできない、嘔吐、無気力、過敏な反応、筋肉のこわばり、発達の遅れといった症状が現れます。治療が遅れると、発作や昏睡に進行し、最終的には命に関わる可能性があります。診断は新生児スクリーニングによって行われ、血液中の分岐鎖アミノ酸やその代謝産物の異常な増加が確認されます。
古典型以外にも、症状の現れ方が異なるいくつかのタイプがあります。「中間型MSUD」は、酵素の働きが部分的に残っているため、血中の分岐鎖アミノ酸の濃度が慢性的に高くなります。症状の進行は緩やかですが、発達の遅れや知的障害が徐々に現れることが多く、診断までに時間がかかる場合があります。「間欠型MSUD」は、通常は正常な代謝機能を持つものの、感染症や断食といった体に強い負荷がかかる状況で急性代謝異常を起こします。この発作時には古典型MSUDと同様の症状が現れますが、適切な食事管理によって症状の発生を抑えることができます。
また、「チアミン反応性MSUD」は、ビタミンB1(チアミン)の高用量投与によって酵素の残存機能が改善するタイプです。このタイプの患者は、分岐鎖アミノ酸の摂取制限が必要な場合もありますが、チアミンを補充することで代謝異常のリスクを軽減し、予後を改善できるとされています。さらに、「E3欠損型MSUD」と呼ばれるタイプは、DLD遺伝子の変異が原因で発症します。この遺伝子はBCKADのほか、ピルビン酸脱水素酵素やα-ケトグルタル酸脱水素酵素といった複数の酵素複合体の構成要素を作る働きを持っています。E3欠損型MSUDでは、これらの酵素の機能が大きく低下し、乳酸アシドーシス(血液中の乳酸の異常な蓄積)や発達の遅れなど、重篤な代謝異常が起こることが特徴です。
MSUDは「常染色体劣性遺伝形式」をとる病気であり、発症には両親からそれぞれ1つずつ異常な遺伝子を受け継ぐ必要があります。一方、片方の遺伝子のみが変異している保因者(キャリア)は通常、症状を示しませんが、子どもに病気を伝える可能性があります。特定の民族集団では、保因者の割合が高いため、MSUDの発症率が高くなることが知られています。
MSUDの治療の基本は、生涯にわたる厳格な食事管理です。具体的には、分岐鎖アミノ酸の摂取を制限し、特殊な医療用ミルクや栄養補助食品を利用して必要な栄養素を補います。急性代謝異常が発生した場合には、静脈からのブドウ糖補給やアミノ酸を含まない輸液の投与といった緊急対応が必要になります。さらに、重症の患者では肝移植が治療の選択肢となることがあります。正常な機能を持つ肝臓を移植することで、分岐鎖アミノ酸の代謝が改善し、代謝異常によるリスクが大幅に軽減されるため、根治的な治療として期待されています。
新生児スクリーニングの発展や食事療法の進歩により、MSUDの管理は大きく向上しましたが、依然として厳格な管理と継続的な医療的監視が求められる病気です。適切な治療を受けなければ、蓄積した有害な代謝産物が脳に深刻なダメージを与え、発作や昏睡を引き起こし、最終的には命に関わることもあります。しかし、早期に診断し、適切な管理を行うことで、患者の健康状態を維持し、比較的安定した生活を送ることが可能です。
ジヒドロリポアミドデヒドロゲナーゼ欠損症(Dihydrolipoamide Dehydrogenase Deficiency: DLD Deficiency)
ジヒドロリポアミドデヒドロゲナーゼ欠損症(Dihydrolipoamide Dehydrogenase Deficiency: DLDD、E3欠損症とも呼ばれる)は、エネルギー代謝に必要な重要な酵素の働きを妨げる、まれな遺伝性疾患です。この疾患では、分岐鎖α-ケト酸デヒドロゲナーゼ複合体(BCKDC)、ピルビン酸デヒドロゲナーゼ複合体(PDC)、α-ケトグルタル酸デヒドロゲナーゼ複合体(KGDC)といった複数の酵素が影響を受けます。これらの酵素には共通の成分であるジヒドロリポアミドデヒドロゲナーゼ(DLD)が含まれており、この酵素が機能しないと酸化代謝がうまく行われず、代謝や神経系に多様な症状が現れます。
DLDDの症状は非常に幅広く、乳児期に重症化するケースから、比較的軽度で成人期に発症するケースまでさまざまです。最も一般的で深刻な症状は乳児期に現れ、筋肉の緊張が低下する低緊張や、乳酸が体内に蓄積する乳酸アシドーシス、そして代謝性の危機(体内のバランスが崩れ、急激に症状が悪化する状態)が特徴です。感染症や絶食、身体的ストレスなどによって症状が悪化し、多くの乳児は最初の代謝性危機を乗り越えられずに亡くなります。生存した場合でも、発達の遅れや知的障害、筋肉のこわばり(痙縮)、運動失調、てんかんなどの神経障害が見られることが多く、成長障害を伴う場合もあります。
この疾患は、神経系ではなく肝臓に主に影響を与える場合もあります。この肝型は、新生児期から成人期まで、どの時期にも発症する可能性があります。典型的な症状としては、まず吐き気や嘔吐が見られ、その後、肝性脳症(意識障害を伴う肝機能の低下)、凝固障害(血が固まりにくくなる状態)、肝腫大(肝臓の肥大)が起こることが多いです。急性の代謝性エピソードでは、血液中の乳酸やアンモニアの濃度が上昇するのが特徴ですが、発作後に肝機能が回復するケースもあります。しかし、成人期に発症した場合でも、肝不全により命に関わることがあります。
さらに、まれなケースとして、DLDDが主に筋肉に影響を与える型もあります。この型では、運動中の筋肉のけいれんや筋力低下が見られ、血中クレアチンキナーゼ(CK)の値が高くなることが特徴です。過去には、重度の運動不耐症を示した患者が、リボフラビン(ビタミンB2)の補給によって改善した例が報告されています。
DLDDの治療では、急性の代謝性危機の管理と、長期的な合併症の予防が重要になります。特に、早期に発症する神経型の患者に対しては、タンパク質の摂取量を適切に管理し、代謝のバランスを保つことが求められます。アミノ酸のバランスが乱れている場合には、分岐鎖アミノ酸(BCAA)を含まない特殊なミルクを使用することもあります。また、ケトン食や高脂肪食が治療法として検討されることがあります。ジクロロ酢酸(DCA)の投与が試されることもありますが、この薬剤には末梢神経障害のリスクがあるため、慎重な管理が必要です。食事療法に加え、摂食障害が続く場合には胃瘻(胃に直接栄養を送る管)の設置が検討されます。さらに、発達の遅れ、心機能障害、視覚障害に対しても適切な対応が必要です。
急性の代謝失調が起こった場合、速やかな入院治療が必要となります。この際、ブドウ糖の静脈投与(D10、年齢に応じた電解質を含む)により、体内のエネルギー不足を補い、分解代謝を抑制します。また、重度のアシドーシス(血液が酸性に傾く状態)には、重炭酸ナトリウムの投与が検討されます。必要に応じて、腎代替療法が行われる場合もあります。タンパク質の摂取量は慎重に調整され、特にロイシンの蓄積を防ぐために、イソロイシンやバリンの補給が行われることがあります。てんかん発作については、標準的な治療が適用されます。
現時点では、DLDDによる急性代謝性危機を完全に防ぐ方法は確立されていません。そのため、定期的な経過観察が重要になります。成長や栄養状態のチェックを定期的に行い、乳幼児期には頻繁な血液検査を実施することが推奨されます。肝機能検査や心エコー検査、眼科検診も必要に応じて行われます。DCAを使用している場合には、末梢神経障害の兆候を慎重に観察する必要があります。
急性代謝性危機のリスクを減らすために、絶食や極端な食事制限を避けることが推奨されます。また、肝臓に負担をかける薬剤の使用は控えるべきです。遺伝的リスクがある家族に対しては、早期の遺伝子検査や生化学的スクリーニングが推奨されます。
DLDDは常染色体劣性遺伝形式で遺伝するため、患者は両親からそれぞれ1つずつDLD遺伝子の変異を受け継いでいます。兄弟姉妹の発症確率は25%、保因者である確率は50%、正常な遺伝子を持つ確率は25%です。保因者の検査や、遺伝子変異が特定されている場合には出生前診断も可能です。
DLDDは重篤な疾患ですが、早期診断と適切な管理により、患者の予後を改善する可能性があります。
メチルマロン酸血症(Methylmalonic acidemia)
| REPRESENTATIVEDISEASES (JA) | メチルマロン酸血症(メチルマロン酸CoAムターゼ欠損症); メチルマロン酸血症、cblA型 |
|---|---|
| REPRESENTATIVEDISEASES (EN) | Methylmalonic acidemia due to methylmalonic-CoA mutase deficiency; Methylmalonic acidemia, cblA type |
| IMPLICATEDGENES | ABCD4, ACSF3, ALDH6A1, CD320, HCFC1, LMBRD1, MCEE, MLYCD, MMAA, MMAB, MMACHC, MMADHC, MMUT, MTR, MTRR, SUCLA2, SUCLG1 |
メチルマロン酸血症(メチルマロン酸CoAムターゼ欠損症)(Methylmalonic acidemia due to methylmalonic-CoA mutase deficiency)
メチルマロン酸血症(MMA)は、遺伝性の代謝異常の一種で、体内で特定のタンパク質や脂肪の成分を適切に分解できなくなる疾患です。この代謝の異常により、有害な物質、特にメチルマロン酸が血液や尿、その他の体液に蓄積します。原因は代謝に関与する酵素の働きを損なう遺伝子変異であり、特にメチルマロニルCoAムターゼという酵素の機能不全が一般的です。MMAは「常染色体劣性遺伝」という形式で遺伝し、発症には両親それぞれから変異した遺伝子を1つずつ受け継ぐ必要があります。
MMAの症状は通常、生後まもなく現れますが、その重症度は人によって異なります。軽度の症状で済む人もいれば、命に関わる合併症を引き起こす場合もあります。主な症状としては、授乳の困難、頻繁な嘔吐、脱水、筋力の低下(筋緊張低下)、極度の疲労感(嗜眠)、発達の遅れ、体重や身長の成長不良などが挙げられます。場合によっては、肝臓が腫れる(肝腫大)こともあります。また、MMAでは「代謝クリーゼ」と呼ばれる急性の代謝障害が繰り返し発生することがあり、これは血液中の酸が過剰に増える「代謝性アシドーシス」を伴います。代謝クリーゼの際には、呼吸困難、意識の混乱、さらには昏睡状態に陥ることもあります。適切な治療が行われなければ、生命の危険を伴うこともあります。
MMAの長期的な影響としては、知的発達の遅れ、運動障害、慢性腎疾患、膵炎(すいえん)などが挙げられます。また、一部の人は食事の摂取が困難になり、特別な栄養管理が必要になることもあります。MMAは単独で発症することが多いですが、場合によってはアミノ酸代謝に影響を与える「ホモシスチン尿症」を伴うこともあります。
MMAには稀なタイプも存在し、その一つにメチルマロニルCoAエピメラーゼの欠損によるものがあります。このタイプでは、体液中のメチルマロン酸の濃度が慢性的に上昇しますが、症状の程度は軽度から中等度にとどまることが多いです。具体的には、嘔吐、脱水、意識の混乱、幻覚などの急性の代謝障害がみられることもあれば、軽度の神経症状にとどまる場合もあります。中には、特に目立った症状がない人もいます。
MMAの診断は、新生児スクリーニング検査、遺伝子検査、血液や尿中のメチルマロン酸濃度を測定する生化学的検査によって確定されます。治療としては、特定のタンパク質の摂取を制限する食事療法や、ビタミンB12の補充(この治療に反応するタイプのMMAの場合)が行われます。また、肝臓や腎臓の移植が、代謝機能の改善を目的として選択されることもあります。MMAは生涯にわたり管理が必要な疾患であり、代謝クリーゼの予防や合併症の対策のために、継続的な医療的フォローが欠かせません。
メチルマロン酸血症、cblA型(Methylmalonic acidemia, cblA type)
メチルマロン酸血症(MMA)のcblA型は、特定のタンパク質や脂肪を適切に代謝する能力に影響を及ぼす、まれな遺伝性の代謝疾患です。この疾患は、メチルマロン酸が体液や組織内に過剰に蓄積することで毒性を引き起こすメチルマロン酸血症(またはメチルマロン酸尿症)の一種であり、特に分岐鎖アミノ酸(BCAA)の代謝異常を伴うことから、有機酸代謝異常の一つとされています。
cblA型MMAは、第4染色体の4q31にあるMMAA遺伝子の変異によって発症します。この遺伝子は、アデノシルコバラミン(AdoCbl)と呼ばれる活性型ビタミンB12の合成に関与し、細胞の正常な代謝に不可欠な役割を果たしています。本疾患は常染色体劣性遺伝の形式をとるため、発症には両親それぞれから変異した遺伝子を1つずつ受け継ぐ必要があります。cblA型MMAの特徴は、ビタミンB12療法に反応する点であり、同じMMAの一種であるmut型(MUT遺伝子の変異によるもの)とは異なります。mut型MMAはビタミンB12の投与に反応しないことが多く、より重篤な経過をたどる傾向があります。
症状やその重症度は個人によって異なりますが、一般的な症状として、哺乳不良、嘔吐、発達の遅れ、成長障害などがあり、特に乳児期に現れやすいとされています。治療を行わずに放置すると、有害な代謝産物が体内に蓄積し、代謝性クリーゼ(急性代謝失調)、神経障害、腎機能障害などの重篤な合併症を引き起こし、場合によっては生命を脅かす急性代謝不全につながることもあります。
このため、早期診断と治療が極めて重要です。新生児マススクリーニング検査でプロピオニルカルニチン(C3)の上昇が確認された場合、診断が確定するまでの間も速やかに代謝管理を開始することが推奨されます。cblA型MMAの治療には、代謝疾患に詳しい専門医チームによる多角的なアプローチが求められます。治療の基本方針として、ビタミンB12に反応する患者には適切な補充療法を行うこと、メチルマロン酸の蓄積を促す特定のアミノ酸(プロピオン酸を生成するアミノ酸)を制限するためにタンパク質の摂取を管理すること、そしてエネルギー不足による代謝の悪化を防ぐために十分なカロリー摂取を確保することが挙げられます。また、哺乳や食事が難しい場合、頻回の嘔吐や成長障害がある場合には、栄養管理やカルニチン補充が必要になることもあります。さらに、腸内細菌によるプロピオン酸の過剰産生を抑制するための対策や、感染症や代謝ストレス時に急性代謝失調を防ぐための緊急代謝サポートも重要です。
病状が不安定で代謝のコントロールが難しい場合や、腎機能の悪化が進行している場合には、肝移植や腎移植が治療の選択肢となることもあります。これにより代謝の安定が期待されるものの、根本的な治療とはならないため、慎重な判断が必要です。
長期的な管理には、定期的な医学的フォローアップが欠かせません。代謝専門医や管理栄養士による診察を受けることが推奨され、血液や尿の検査でアミノ酸濃度、メチルマロン酸値、アシルカルニチンプロファイル、カルニチン濃度などを定期的に測定し、全体的な代謝バランスを確認します。腎機能は最低でも年に1回の検査が必要で、肝疾患の有無、発達の進行状況、運動障害の兆候についても継続的に評価する必要があります。また、視神経萎縮や聴覚障害が発症する可能性があるため、眼科や耳鼻科での定期検査も重要です。
代謝の急激な悪化を防ぐために、長時間の絶食や高タンパク食を避け、強い身体的・精神的ストレスをできるだけ軽減することが推奨されます。また、腎毒性のある薬剤や、QTc延長を引き起こす可能性がある薬剤の使用には慎重な対応が必要です。妊娠中の管理については、ビタミンB12に反応するMMA胎児を持つ母親にビタミンB12を投与することで、母体の尿中メチルマロン酸排泄量が減少する可能性があるとの報告もありますが、さらなる研究が求められています。
適切な治療と管理を行うことで、cblA型MMAの患者は代謝の安定化と長期的な健康維持が期待できます。ただし、病状の進行を抑えるためには、早期の介入、継続的な医療管理、そして個々の状態に応じた治療計画の策定が不可欠です。
ペルオキシソーム障害(Peroxisomal disorders)
| REPRESENTATIVEDISEASES (JA) | 副腎白質ジストロフィー、ペルオキシソーム生合成障害 1A、2A、2B(ゼルウェガー症候群) |
|---|---|
| REPRESENTATIVEDISEASES (EN) | Adrenoleukodystrophy; Peroxisome biogenesis disorder 1A, 2A, 2B (Zellweger syndrome) |
| IMPLICATEDGENES | ABCD1, ACOX1, AGPS, AMACR, HSD17B4, PEX1, PEX2, PEX3, PEX5, PEX6, PEX7, PEX10, PEX11B, PEX12, PEX13, PEX14, PEX16, PEX19, PEX26, PHYH, SCP2 |
副腎白質ジストロフィー(Adrenoleukodystrophy)
副腎白質ジストロフィー(Adrenoleukodystrophy, ALD)は、神経系と副腎に影響を及ぼす遺伝性の希少疾患です。この病気は、ABCD1遺伝子の変異によって引き起こされ、非常に長鎖の脂肪酸(Very Long-Chain Fatty Acids, VLCFA)が体内のさまざまな組織、特に脳や副腎皮質に蓄積することが特徴です。この蓄積により細胞の正常な機能が妨げられ、神経を保護するミエリンが徐々に破壊されるほか、副腎の機能が低下し、ホルモン分泌の異常が生じます。
ALDはX染色体に関連する遺伝病であるため、主に男性に症状が現れます。一方、女性は保因者となることが多く、重篤な症状を発症することはまれですが、加齢とともに軽度の神経症状を示すことがあります。ALDにはいくつかの異なるタイプがあり、それぞれ特徴や重症度が異なります。
最も重症なタイプである「小児脳型ALD」は、主に4歳から10歳の間に発症します。初期症状として学習の遅れや注意力の低下などがみられますが、その後、視力の低下、嚥下障害、運動機能の低下、攻撃的な行動、言語障害が進行し、最終的には意思疎通や身体の自由な動きが困難になります。治療を受けない場合、数年以内に重度の障害を抱え、早期に生命を脅かすことが多い病型です。
成人期以降に発症するタイプとして「副腎脊髄ニューロパチー(Adrenomyeloneuropathy, AMN)」があります。主に20代から40代にかけて症状が現れ、脚の筋力低下やこわばりが徐々に進行し、歩行が困難になります。また、排尿障害や性機能の低下、行動の変化、認知機能の低下を伴うこともあります。AMNは小児脳型ALDに比べて進行が遅いものの、多くの患者が最終的に重度の神経障害を抱えることになります。さらに、AMNの患者の多くは副腎機能不全を併発しており、生涯にわたりホルモン補充療法が必要になります。
一部の患者では、副腎機能不全のみが症状として現れることもあります。このタイプは「神経症状を伴わないアジソン病」とも呼ばれ、幼少期から成人期のいずれでも発症する可能性があります。しかし、この状態の患者も後に脳型ALDやAMNを発症することがあるため、早期の診断と適切な経過観察が重要です。
ALDには、明らかな症状がない「無症候性」のケースもあります。この場合、血液検査でVLCFA値が高い、あるいは脳の画像検査で初期の異常が見られるものの、日常生活に支障が出るような症状はありません。しかし、こうした人々の一部は将来的に症状を発症する可能性があり、継続的な経過観察が推奨されます。
まれに、思春期や若年成人期に精神症状や認知症のような症状を伴う特殊なケースも報告されています。同じ家族内でも異なるタイプのALDが発症することがあり、その原因はまだ十分に解明されていません。
ALDの診断には、血液検査でVLCFAの濃度を測定する方法が一般的に用いられます。ほぼすべての男性患者でVLCFA値の上昇が確認されるため、診断の手がかりとなります。確定診断には遺伝子検査を行い、ABCD1遺伝子の変異を特定します。特に小児脳型ALDでは早期診断が非常に重要であり、骨髄移植や幹細胞移植などの治療は、病気の進行が始まる前の段階で最も効果を発揮します。また、副腎機能不全の患者にはホルモン補充療法が不可欠です。
現在のところ、ALDの根本的な治療法は確立されておらず、主に病気の進行を遅らせたり、症状を和らげることが治療の中心となっています。しかし、近年では遺伝子治療や食事療法などの研究が進められており、将来的にはより効果的な治療法が開発されることが期待されています。
ゼルウェガー症候群(Zellweger syndrome)
Zellweger症候群は、極めてまれで重篤な遺伝性疾患の一つであり、Zellwegerスペクトラムと呼ばれる病気の中でも最も重いタイプに分類されます。この疾患は、細胞内に存在するペルオキシソームという構造の形成異常によって引き起こされます。ペルオキシソームは、脂肪酸の分解や有害物質の処理など、細胞の代謝において重要な役割を果たしています。しかし、Zellweger症候群ではペルオキシソームが完全に欠損しているか、その機能が著しく低下しているため、全身のさまざまな臓器や組織に深刻な影響を及ぼします。
この病気の症状は、生まれてすぐ、あるいは生後間もなく現れることが多く、特に筋肉の緊張が極端に低下する「筋緊張低下(低緊張)」が特徴的です。そのため、赤ちゃんがうまく母乳やミルクを飲めないことがあり、けいれんを起こすこともあります。また、聴覚や視覚の障害がみられるほか、額が広い、大泉門(頭蓋骨のつなぎ目)が通常より大きい、目が離れているなどの特徴的な顔立ちを示すことがあります。さらに、関節が硬くなって動かしにくくなる関節拘縮や、軟骨や骨の形成異常(点状軟骨異形成症)といった骨格の異常もみられることがあります。
Zellweger症候群は見た目の特徴だけでなく、内臓にも深刻な影響を及ぼします。肝機能の障害によって黄疸や血液の凝固異常が起こることがあり、心臓や腎臓にも異常が生じることがあります。また、脳の発達においても大きな影響があり、神経細胞が適切な位置へ移動する過程(神経移動)が正常に行われないため、発達の遅れや重度の知的障害を伴うことが多くなります。
この疾患は、常染色体劣性遺伝という遺伝形式で受け継がれます。つまり、発症するためには両親からそれぞれ変異した遺伝子を受け継ぐ必要があります。Zellweger症候群の原因となる遺伝子は少なくとも12種類以上が確認されており、その中でもPEX1遺伝子の変異が最も一般的です。
現在のところ、Zellweger症候群に対する根本的な治療法はなく、症状を和らげることを目的とした対症療法が中心となります。たとえば、哺乳が難しい場合には、胃に直接栄養を送るための胃瘻(いろう)を造設し、必要な栄養を補給します。聴覚障害には補聴器を使用し、白内障がある場合には手術で除去することが検討されます。視力の低下には眼鏡の使用が推奨されることがあります。また、肝機能の低下に対しては脂溶性ビタミンやコール酸の補充が行われることがあり、けいれん発作には抗てんかん薬が用いられます。発達の遅れがある場合は、早期からのリハビリテーションや発達支援を行うことが重要です。
また、合併症に対する治療も必要になることがあります。たとえば、副腎機能が低下する副腎機能不全がみられる場合には、副腎ホルモン補充療法を行います。骨密度の低下(骨減少症)を防ぐために、ビタミンDやビスホスホネート製剤の使用が検討されることもあります。歯のエナメル質が正常に形成されない「エナメル質形成不全」がある場合には、専門的な歯科治療が必要となります。腎臓にシュウ酸カルシウム結石ができることがあり、その場合には水分摂取を適切に管理したり、体外衝撃波結石破砕術(リトトリプシー)や外科手術を検討することもあります。
Zellweger症候群の進行を適切に管理するためには、定期的な医療的フォローが欠かせません。成長や栄養状態は診察のたびに確認し、聴覚や視覚の評価は毎年行うことが推奨されます。肝機能や血液の凝固機能を定期的に検査し、超音波検査やフィブロスキャンを用いて肝臓の状態を評価することが重要です。脳のMRI検査を行い、白質(神経の伝達を担う部分)の変化を観察することで、認知機能や運動能力の低下の原因を把握することができます。また、発作の頻度や発達の進行状況を確認し、教育的な支援が適切に行われているかを定期的に見直すことも求められます。内分泌機能の異常を早期に発見するために、副腎皮質刺激ホルモン(ACTH)やコルチゾールの測定を1歳までに開始し、その後も毎年継続的に検査することが望ましいとされています。歯科検診は6か月ごとに受けることが勧められ、腎機能を評価するために尿中シュウ酸の測定を定期的に行い、必要に応じて腎臓の画像診断を実施することも検討されます。さらに、感染症予防のために、毎年インフルエンザワクチンやRSウイルス(呼吸器合胞体ウイルス)ワクチンを接種することが推奨されます。
Zellweger症候群は進行性の病気であり、生命を脅かす疾患であるため、患者本人だけでなく、その家族に対するサポートも非常に重要です。診察時には、家族が必要とする支援についても確認し、適切なリソースやカウンセリングを提供できるようにすることが大切です。根本的な治療法は存在しませんが、適切な医療ケアと支援を受けることで、患者の生活の質をできる限り向上させ、快適に過ごせるようにすることが可能です。
尿素サイクル異常症(Urea cycle disorders)
| REPRESENTATIVEDISEASES (JA) | アルギニン血症、シトルリン血症 |
|---|---|
| REPRESENTATIVEDISEASES (EN) | Argininemia, Citrullinemia |
| IMPLICATEDGENES | ARG1, ASL, ASS1, CPS1, NAGS, OTC, SLC25A13, SLC25A15 |
アルギニン血症(Argininemia)
アルギナーゼ欠損症(アルギニン血症)は、体内でアルギニンというアミノ酸を適切に分解できなくなる、まれな遺伝性の代謝異常症です。アルギニンはタンパク質の代謝に欠かせない成分ですが、この病気ではその処理がうまくいかず、血液中にアルギニンやアンモニアが徐々に蓄積していきます。この疾患は尿素サイクル異常症(尿素回路障害)の一つであり、体内のアンモニア排出機能が損なわれることで、神経や発達に影響を及ぼすさまざまな症状が現れます。
アルギナーゼ欠損症の原因は、ARG1遺伝子の変異です。この遺伝子はアルギナーゼという酵素の働きを調整しており、アルギナーゼはアルギニンを尿素とオルニチンに分解する役割を担っています。しかし、遺伝子に異常があるとこの酵素の働きが弱まり、アルギニンの分解が滞ることで血液や脳脊髄液中のアルギニン濃度が上昇します。また、時折、アンモニアが血液中に過剰に蓄積する高アンモニア血症の発作が起こることがあります。アンモニアは神経に対する強い毒性を持つため、その蓄積が続くと脳や神経の働きに深刻な影響を及ぼします。
この疾患の症状は通常、生後数年間は目立ちにくいものの、多くの場合、3歳頃から現れ始めます。初期症状のひとつとして、足を中心とした筋肉のこわばりがみられ、歩行が難しくなることがあります。このこわばりは、筋肉の緊張が異常に高まる痙縮(けいしゅく)によるものです。また、成長の遅れや発達の遅延が見られることがあり、それまでできていた歩行や言葉の発達が徐々に失われることもあります。適切な治療を受けない場合、筋肉のこわばりが悪化し、最終的には歩行が困難になったり、知的発達の遅れが深刻化したりする可能性があります。その他にも、けいれん(発作)、震え(振戦)、バランス感覚の障害(運動失調)、過剰な反射反応などが現れることがあります。一方で、症状の程度や発症時期には個人差があり、一部の患者では比較的軽度であったり、発症が遅れたりすることもあります。
また、高アンモニア血症の発作は、感染症、空腹状態(絶食)、または高タンパク質の食事を摂取した際に誘発されることがあります。発作が起こると、強い不機嫌や食欲不振、嘔吐などの症状がみられ、アンモニアの毒性による強い不快感が生じることがあります。
アルギナーゼ欠損症は、常染色体劣性遺伝形式で遺伝する病気であり、発症するためには両親からそれぞれ変異したARG1遺伝子を受け継ぐ必要があります。診断は血液や脳脊髄液の検査でアルギニン濃度の異常を確認するほか、遺伝子検査によりARG1遺伝子の変異を特定することで確定されます。
治療の基本は、アルギニンやアンモニアの蓄積を抑えるための食事療法や代謝管理です。特に低タンパク質の食事を徹底することが重要であり、専門の管理栄養士の指導のもとで栄養バランスを適切に調整することが推奨されます。また、一部の患者には、窒素の排出を助ける薬剤(窒素スカベンジャー薬)を使用することで、高アンモニア血症のリスクを軽減する治療が行われることもあります。早期診断と継続的な管理により、神経への影響を最小限に抑え、患者の生活の質を向上させることができます。
現時点ではアルギナーゼ欠損症を根本的に治す治療法は存在しませんが、早期の介入と適切な医療管理により症状の進行を抑え、患者ができる限り自立した生活を送ることが可能になります。
シトルリン血症(Citrullinemia)
シトルリン血症は、体内で窒素を適切に処理する機能に異常が生じる遺伝性の希少疾患であり、その結果、血液中にアンモニアやその他の有害な物質が蓄積します。この病気は「尿素サイクル異常症」に分類され、常染色体劣性遺伝の形式をとります。つまり、発症するためには両親からそれぞれ変異した遺伝子を受け継ぐ必要があります。シトルリン血症にはいくつかのタイプがあり、発症時期や症状の現れ方は異なります。
シトルリン血症には主に2つのタイプがあり、それぞれ異なる遺伝子の変異によって引き起こされます。シトルリン血症I型(クラシックシトルリン血症とも呼ばれる)は、比較的一般的なタイプで、多くの場合、生後数日以内に発症します。新生児は出生直後には健康に見えますが、血液中のアンモニア濃度が上昇すると、強い眠気(傾眠)、哺乳不良、嘔吐、けいれん、意識障害といった症状が現れます。適切な治療が行われない場合、肝機能障害や重篤な高アンモニア血症を引き起こし、生命の危険を伴うことがあります。I型には、まれに幼児期や成人になってから発症する軽症型もあり、この場合、激しい頭痛、視界の一部が暗くなる(暗点)、バランス感覚や運動機能の障害(運動失調)、断続的な倦怠感といった症状が見られます。また、I型シトルリン血症の原因となる遺伝子変異を持っていても、一生涯にわたって症状が現れない人もいます。
一方、シトルリン血症II型は主に神経系に影響を及ぼし、成人になってから発症することが多い病気です。症状には、混乱、記憶障害、攻撃的または過敏な行動の変化、多動、けいれん、さらには昏睡状態が含まれます。また、II型の患者は、タンパク質や脂肪を多く含む食品を好み、炭水化物を多く含む食事を避ける傾向があるといわれています。発症は、感染症、アルコール摂取、手術、特定の薬の使用などがきっかけとなることがあり、適切な治療が行われない場合には生命の危険を伴うこともあります。
II型シトルリン血症には、新生児期に発症するタイプもあり、これはシトリン欠損症と関連しています。シトリン欠損症を持つ新生児の一部は、「シトリン欠損による新生児肝内胆汁うっ滞症(NICCD; neonatal intrahepatic cholestasis caused by citrin deficiency)」と呼ばれる肝疾患を発症します。NICCDは胆汁の流れを妨げ、栄養の代謝に影響を与える病気で、黄疸、体重増加不良、栄養バランスの乱れなどの症状を引き起こします。ただし、多くの場合、生後1年以内に症状は自然に改善します。しかし、一度NICCDの症状が治まったように見えても、その後、成長障害、極度の疲労、特定の食べ物の嗜好(タンパク質や脂肪を好み、炭水化物を避ける傾向)、血中脂質異常といった症状が現れることがあります。この状態は「シトリン欠損による成長不良と脂質異常症(FTTDCD)」と呼ばれます。さらに稀なケースでは、NICCDやFTTDCDの既往がある人が、数年から数十年後にII型シトルリン血症の神経症状を発症することもあります。
シトルリン血症は重篤な合併症を引き起こす可能性がありますが、新生児スクリーニング検査による早期発見と適切な医療管理によって、病状の進行を抑えることが可能です。治療としては、タンパク質の摂取を制限する食事療法、アンモニアを排出するための薬剤の使用、重症の場合は肝移植が検討されることもあります。症状の現れ方には個人差があり、まったく症状が出ない人もいるため、リスクのある人は遺伝子検査や定期的な医療チェックを受けることが重要です。
【出産後テスト】Newborn Testing
はじめに
ヒロクリニックの「出産後テスト」について
ヒロクリニックの「出産後テスト」は、新生児の健康状態を確認するためのスクリーニング検査です。生後に発症する可能性のある19種類の疾患を早期に発見し、適切な対応につなげることを目的としています。すべての新生児に推奨されますが、特に以下のような症状が見られる場合は、より重要な検査となります。
検査を検討すべき主な症状
-
呼吸の異常
- 息が速く浅い
- 呼吸のリズムが不規則
- 一定時間、呼吸が止まる(無呼吸)
-
授乳の問題
- 母乳やミルクを十分に吸えない
- 飲む力が弱い
- 成長が遅れている
-
黄疸(おうだん)
- 皮膚や白目が黄色くなる症状
- 多くは自然に改善するが、医療的対応が必要な場合もある
-
低体重や成長の遅れ
- 出生時の体重が低い場合
- 体重がなかなか増えない
-
体温の異常
- 通常より低い(低体温症)
- 高すぎる(高体温症)
-
その他の気になる症状
- 極端に眠りがちで反応が鈍い(嗜眠〈しみん〉)
- けいれん発作を起こす
検査を特に推奨するケース
以下のような赤ちゃんには、特にこの検査が推奨されます。
- 早産で生まれた
- 分娩時に合併症があった
- 先天的な異常がある
- 母親が妊娠中に感染症にかかった
- 環境汚染物質に長期間さらされた可能性がある
なぜ早期検査が重要なのか
内分泌疾患、代謝疾患、免疫疾患などは、発症頻度は低いものの、すべてを含めると3,000~3,500人に1人の割合で見られます。こうした疾患は、新生児期には見た目が健康に見えることが多く、見逃されやすい特徴があります。
しかし、適切な診断と治療が行われなければ、成長とともに学習障害や頻繁な体調不良などの問題が現れる可能性があります。重症化すると細胞や臓器の損傷が進み、回復が難しくなることもあり、最悪の場合、生命に関わる事態へと進行することも考えられます。
早期発見ができれば、リスクを大幅に軽減できる可能性があります。
検査を通じて適切な治療を早期に開始することで、赤ちゃんの健康な未来を支えることができます。
新生児スクリーニングとは
新生児スクリーニング(Newborn Screening, NBS)は、赤ちゃんの健康を守るための重要な予防医療のひとつです。生まれたばかりの赤ちゃんはまだ症状を示していなくても、NBSによって深刻ながらも治療可能な病気を早期に発見することができます。これにより、適切な治療を迅速に開始し、健康状態の改善につなげることが可能になります。
このスクリーニングは1960年代に始まり、当初はフェニルケトン尿症(Phenylketonuria, PKU)のみを対象としていました。しかし、その後の技術革新により、スクリーニングの対象疾患は大幅に増加しました。近年では、遺伝子解析技術の発展によって、より多くの疾患を検出できるようになっています。
希少疾患とゲノム新生児スクリーニング(gNBS)
希少疾患は乳幼児の病気や死亡の主な原因のひとつですが、早期に発見できれば治療可能な場合もあります。従来の新生児スクリーニング(Traditional Newborn Screening, tNBS)は、幼少期に重篤な症状を示しながらも治療が可能な疾患に焦点を当てていました。しかし、新しい技術の進歩により、生まれてすぐに数百種類の遺伝性疾患を検査できるようになりつつあります。
この拡張されたスクリーニングは「ゲノム新生児スクリーニング(Genomic Newborn Screening, gNBS)」と呼ばれ、より多くの疾患を早期に診断し、健康な成長を支援することが期待されています。さらに、赤ちゃんの遺伝情報を保存しておくことで、将来新たな健康問題が発生した際に再解析することも可能になります。
新生児スクリーニングの技術的進歩
新生児スクリーニングの技術は、近年飛躍的に向上しています。かつてはごく限られた疾患しか検査できませんでしたが、医療技術の発展に伴い、対象疾患の数が増加しました。
特に、タンデム質量分析(Tandem Mass Spectrometry, MS/MS)の導入は大きな進歩でした。この技術により、わずかな血液サンプルから多数の代謝異常疾患を同時に検査できるようになりました。さらに、近年では遺伝子解析技術の発展により、赤ちゃんの全遺伝情報を解析し、病気に関連する遺伝子変異を特定することが可能になっています。この方法は迅速かつ高精度であり、費用も徐々に低下しています。
しかし、技術の進歩には慎重な検討も必要です。スクリーニングの対象を拡大することで、医療的・倫理的・社会的な課題が生じる可能性があります。
スクリーニングの倫理的・社会的課題
新生児スクリーニングの拡大に伴い、さまざまな倫理的・社会的課題が指摘されています。その一つとして、遺伝子の変異が見つかった場合でも、それが将来的に病気を引き起こすかどうかが不明なケースがあります。このような場合、確実な診断ができないまま家族に不安を与える可能性があります。
また、遺伝情報の取り扱いに関しては、プライバシーの保護や情報の悪用リスクが懸念されています。例えば、遺伝情報が差別や偏見につながる可能性があるため、適切な管理が求められます。さらに、診断基準が国ごとに異なることから、スクリーニング結果の比較が難しいという問題もあります。これにより、国際的な統一基準の確立が求められる状況です。
こうした課題を踏まえると、保護者が検査内容を十分に理解し、適切な判断を下せるようにすることが重要です。そのためには、インフォームド・コンセント、つまり十分な説明を受けた上での同意の確保が不可欠となります。
治療可能な疾患の増加と早期発見の重要性
治療可能な希少疾患の数は年々増加しています。2021年時点では、1,400種類以上の代謝異常疾患のうち116種類に治療法が確立されており、新たな治療も次々と開発されています。
例えば、脊髄性筋萎縮症(Spinal Muscular Atrophy, SMA)やメタクロマチック白質ジストロフィー(Metachromatic Leukodystrophy, MLD)など、かつて治療が困難とされていた疾患にも有効な治療法が登場し、早期発見の重要性がますます高まっています。
しかし、検査項目が多ければ多いほど良いというわけでもありません。すべての遺伝的変異が病気につながるわけではなく、不必要な不安や医療介入の増加につながる可能性があります。そのため、スクリーニングプログラムは、家族に有益な情報を提供しながらも、不安を最小限に抑えるよう設計されるべきです。
もっと詳しく知りたい方へ
表1. 本検査に対応されている疾患
| s/n | Disorder | 疾患名 | 遺伝子 | 遺伝形式 |
|---|---|---|---|---|
| 1 | Adrenogenital syndrome (AGS) | 副腎性器症候群 (AGS) | CYP21A2 | AR |
| 2 | Biotinidase deficiency | ビオチニダーゼ欠損症 | BTD | AR |
| 3 | Carnitine acylcarnitine translocase (CACT) deficiency | カルニチンアシルカルニチントランスロカーゼ (CACT) 欠損症 | SLC25A20 | AR |
| 4 | Carnitine palmitoyltransferase 1A (CPT1A) deficiency | カルニチンパルミトイルトランスフェラーゼ1A (CPT1A) 欠損症 | CPT1A | AR |
| 5 | Carnitine palmitoyltransferase 2 (CPT2) deficiency | カルニチンパルミトイルトランスフェラーゼ2 (CPT2) 欠損症 | CPT2 | AR |
| 6 | Cystic fibrosis (CF) | 嚢胞性線維症 (CF) | CFTR | AR |
| 7 | Galactosemia | ガラクトース血症 | GALT | AR |
| 8 | Glutaric aciduria type 1 (GA1) | グルタル酸血症1型 (GA1) | GCDH | AR |
| 9 | Congenital hypothyroidism | 先天性甲状腺機能低下症 | DUOX2, PAX8, TSHR | AD, AR |
| 10 | Isovaleric acidemia | イソ吉草酸血症 | IVD | AR |
| 11 | Long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency | 長鎖3-ヒドロキシアシルCoA脱水素酵素 (LCHAD) 欠損症 | HADHA, HADHB | AR |
| 12 | Maple syrup urine disease type Ia | メープルシロップ尿症 1a 型 | BCKDHA | AR |
| 13 | Maple syrup urine disease type Ib | メープルシロップ尿症 1b 型 | BCKDHB | AR |
| 14 | Maple syrup urine disease type II | メープルシロップ尿症 2 型 | DBT | AR |
| 15 | Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency | 中鎖アシルCoA脱水素酵素 (MCAD) 欠損症 | ACADM | AR |
| 16 | Phenylketonuria / Hyperphenylalaninemia (PKU / HPA) | フェニルケトン尿症 / 高フェニルアラニン血症 (PKU / HPA) | PAH | AR |
| 17 | Severe combined immunodeficiency (SCID) | 重症複合免疫不全症 (SCID) | 複数の遺伝子 | AR, XLR |
| 18 | Tyrosinemia type 1 | チロシン血症1型 | FAH | AR |
| 19 | Very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency | 超長鎖アシルCoA脱水素酵素 (VLCAD) 欠損症 | ACADVL | AR |
| ※AD: autosomal dominant(常染色体優性)、AR: autosomal recessive(常染色体劣性)、XLR: X-linked recessive(伴性劣性) | ||||
引用文献 & もっと詳しく知りたい方へ
【乳がんと卵巣がんのテスト】
- Presentia BRCA1 / BRCA2 Panel. Retrieved 24 January 2025, from https://medicover-genetics.com/product/presentia-brca1-brca2-panel/
- Kuchenbaecker, K. B., Hopper, J. L., Barnes, D. R., Phillips, K. A., Mooij, T. M., Roos-Blom, M. J., Jervis, S., van Leeuwen, F. E., Milne, R. L., Andrieu, N., et al. (2017). Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA, 317(23), 2402–2416. https://doi.org/10.1001/jama.2017.7112
- Bertozzi, S., Londero, A., Xholli, A., Azioni, G., Di Vora, R., Paudice, M., Bucimazza, I., Cedolini, C., & Cagnacci, A. (2023). Risk-reducing breast and gynecological surgery for BRCA mutation carriers: A narrative review. Journal of Clinical Medicine, 12(4), 1422. https://doi.org/10.3390/jcm12041422
- Petrucelli, N., Daly, M. B., & Pal, T. (1998, updated 2023). BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. In: Adam MP, Feldman J, Mirzaa GM, et al. (Eds.), GeneReviews®. University of Washington, Seattle. https://www.ncbi.nlm.nih.gov/books/NBK1247/
- Fu, X., Tan, W., Song, Q., Pei, H., & Li, J. (2022). BRCA1 and breast cancer: Molecular mechanisms and therapeutic strategies. Frontiers in Cell and Developmental Biology, 10, 813457. https://doi.org/10.3389/fcell.2022.813457
- Zhong, A. X., Chen, Y., & Chen, P.-L. (2023). BRCA1 the versatile defender: Molecular to environmental perspectives. International Journal of Molecular Sciences, 24(18), 14276. https://doi.org/10.3390/ijms241814276
- Werner, H. (2022). BRCA1: An endocrine and metabolic regulator. Frontiers in Endocrinology, 13, 844575. https://doi.org/10.3389/fendo.2022.844575
- Bommer, C., Lupatsch, J., Bürki, N., et al. (2022). Cost–utility analysis of risk-reducing strategies to prevent breast and ovarian cancer in BRCA-mutation carriers in Switzerland. European Journal of Health Economics, 23, 807–821. https://doi.org/10.1007/s10198-021-01396-9
【代謝病のテスト】
- Clarke, S. L., Bowron, A., Gonzalez, I. L., et al. (2013). Barth syndrome. Orphanet Journal of Rare Diseases, 8, 23. https://doi.org/10.1186/1750-1172-8-23
- Jefferies, J. L. (2013). Barth syndrome. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 163(3), 198–205. https://doi.org/10.1002/ajmg.c.31372
- Huizing, M., Dorward, H., Ly, L., Klootwijk, E., Kleta, R., Skovby, F., Pei, W., Feldman, B., Gahl, W. A., & Anikster, Y. (2010). OPA3, mutated in 3-methylglutaconic aciduria type III, encodes two transcripts targeted primarily to mitochondria. Molecular Genetics and Metabolism, 100(2), 149–154. https://doi.org/10.1016/j.ymgme.2010.03.005
- Kleta, R., Skovby, F., Christensen, E., Rosenberg, T., Gahl, W. A., & Anikster, Y. (2002). 3-Methylglutaconic aciduria type III in a non-Iraqi-Jewish kindred: Clinical and molecular findings. Molecular Genetics and Metabolism, 76(3), 201–206. https://doi.org/10.1016/S1096-7192(02)00047-1
- Ojala, T., Polinati, P., Manninen, T., Hiippala, A., Rajantie, J., Karikoski, R., Suomalainen, A., & Tyni, T. (2012). New mutation of mitochondrial DNAJC19 causing dilated and noncompaction cardiomyopathy, anemia, ataxia, and male genital anomalies. Pediatric Research, 72(4), 432–437. https://doi.org/10.1038/pr.2012.92
- 3-Methylglutaconic aciduria type 5 — National Organization for Rare Disorders. (n.d.). Retrieved January 24, 2025, from https://rarediseases.org/mondo-disease/3-methylglutaconic-aciduria-type-5/
- 3-Methylglutaconic aciduria type 5 — NIH Genetic Testing Registry (GTR) — NCBI. (n.d.). Retrieved January 24, 2025, from https://www.ncbi.nlm.nih.gov/gtr/conditions/C1857776/
【出産後テスト】
- Stark, Z., & Scott, R. H. (2023). Genomic newborn screening for rare diseases. Nature Reviews Genetics, 24, 755–766. https://doi.org/10.1038/s41576-023-00621-w
- Remec, Z. I., Trebusak Podkrajsek, K., Repic Lampret, B., Kovac, J., Groselj, U., Tesovnik, T., Battelino, T., & Debeljak, M. (2021). Next-generation sequencing in newborn screening: A review of current state. Frontiers in Genetics, 12, 662254. https://doi.org/10.3389/fgene.2021.662254
- Downie, L., Halliday, J., Lewis, S., & Amor, D. J. (2021). Principles of genomic newborn screening programs: A systematic review. JAMA Network Open, 4(7), e2114336. https://doi.org/10.1001/jamanetworkopen.2021.14336
- Spiekerkoetter, U., Bick, D., Scott, R., Hopkins, H., Krones, T., Gross, E. S., & Bonham, J. R. (2023). Genomic newborn screening: Are we entering a new era of screening? Journal of Inherited Metabolic Disease, 46(5), 778–795. https://doi.org/10.1002/jimd.12650
- Bick, D., Ahmed, A., Deen, D., Ferlini, A., Garnier, N., Kasperaviciute, D., Leblond, M., Pichini, A., Rendon, A., Satija, A., Tuff-Lacey, A., & Scott, R. H. (2022). Newborn screening by genomic sequencing: Opportunities and challenges. International Journal of Neonatal Screening, 8(3), 40. https://doi.org/10.3390/ijns8030040
- La Marca, G., Carling, R. S., Moat, S. J., Yahyaoui, R., Ranieri, E., Bonham, J. R., & Schielen, P. C. J. I. (2023). Current state and innovations in newborn screening: Continuing to do good and avoid harm. International Journal of Neonatal Screening, 9(1), 15. https://doi.org/10.3390/ijns9010015

